Myasthenia gravis (MG) is the most common autoimmune disease affecting neuromuscular junction transmission. MG is characterized by muscle weakness that worsens with activity and fluctuates over the course of the day. Involvement of respiratory musculature can lead to a life-threatening crisis requiring intensive care unit care. Antibody testing is positive in most patients with MG. Treatment of MG includes short-term symptomatic treatment, chronic immunosuppression, surgical intervention, and immunomodulatory therapies for severe disease or crisis. We review advances in 5 areas relevant to diagnosis and management of MG: the role of IV immunoglobulin vs plasmapheresis in myasthenic crisis and severe disease; the clinical characterization of patients with antibodies to muscle-specific tyrosine kinase receptors; old and new investigational treatments; management of MG in pregnancy; and new confirmatory diagnostic tests.
Myasthenia gravis is a long-term neuromuscular disease that leads to varying degrees of skeletal muscle weakness.The most commonly affected muscles are those of the eyes, face, and swallowing. It can result in double vision, drooping eyelids, trouble talking, and trouble walking. Onset can be sudden. Those affected often have a large thymus gland or develop a thymoma.
Types of Myasthenia Gravis
Subtypes of MG are broadly classified as follows
- early-onset MG: age at onset <50 years. Thymic hyperplasia, usually females,
- late-onset MG: age at onset >50 years. Thymic atrophy, mainly males,
- thymoma-associated MG (10%–15%)
- MG with anti-MUSK antibodies,
- ocular MG (oMG): symptoms only affecting extraocular muscles,
- MG with no detectable AChR and muscle-specific tyrosine kinase (MuSK) antibodies
- The most widely utilized classification of MG is the Myasthenia Gravis Foundation of America Clinical Classification
The Myasthenia Gravis Foundation of America Clinical Classification divides MG into 5 main classes and several subclasses :
-
Class I: Any ocular muscle weakness; may have weakness of eye closure; all other muscle strength is normal
-
Class II: Mild weakness affecting other than ocular muscles; may also have ocular muscle weakness of any severity
-
Class IIa: Predominantly affecting limb, axial muscles, or both; may also have lesser involvement of oropharyngeal muscles
-
Class IIb: Predominantly affecting oropharyngeal, respiratory muscles, or both; may also have lesser or equal involvement of limb, axial muscles, or both
-
Class III: Moderate weakness affecting other than ocular muscles; may also have ocular muscle weakness of any severity
-
Class IIIa: Predominantly affecting limb, axial muscles, or both; may also have lesser involvement of oropharyngeal muscles
-
Class IIIb: Predominantly affecting oropharyngeal, respiratory muscles, or both; may also have lesser or equal involvement of limb, axial muscles, or both
-
Class IV: Severe weakness affecting other than ocular muscles; may also have ocular muscle weakness of any severity
-
Class IVa: Predominantly affecting limb, axial muscles, or both; may also have lesser involvement of oropharyngeal muscles
-
Class IVb: Predominantly affecting oropharyngeal, respiratory muscles, or both; may also have lesser or equal involvement of limb, axial muscles, or both; use of a feeding tube without intubation
-
Class V: Defined by the need for intubation, with or without mechanical ventilation, except when used during routine postoperative management
or
The Myasthenia Gravis Foundation of America (MGFA) clinical classification divides MG into 5 main classes and several subclasses [rx]. It is designed to identify subgroups of patients with MG who share distinct clinical features or severity of disease that may indicate different prognoses or responses to therapy. It should not be used to measure outcome and is as follows.
Class I MG is characterized by the following:
-
any ocular muscle weakness.
-
may have weakness of eye closure.
-
all other muscle strengths are normal.
Class II MG is characterized by the following:
-
mild weakness affecting muscles other than ocular muscles,
-
may also have ocular muscle weakness of any severity.
Class IIa MG is characterized by the following:
-
predominantly affecting limb, axial muscles, or both
-
may also have lesser involvement of oropharyngeal muscles.
Class IIb MG is characterized by the following:
-
predominantly affecting oropharyngeal, respiratory muscles, or both,
-
may also have lesser or equal involvement of limb, axial muscles, or both.
Class III MG is characterized by the following:
-
moderate weakness affecting muscles other than ocular muscles,
-
may also have ocular muscle weakness of any severity.
Class IIIa MG is characterized by the following:
-
predominantly affecting limb, axial muscles, or both,
-
may also have lesser involvement of oropharyngeal muscles.
Class IIIb MG is characterized by the following:
-
predominantly affecting oropharyngeal, respiratory muscles, or both,
-
may also have lesser or equal involvement of limb, axial muscles, or both.
Class IV MG is characterized by the following:
-
severe weakness affecting muscles other than ocular muscles,
-
may also have ocular muscle weakness of any severity.
Class IVa MG is characterized by the following:
-
predominantly affecting limb, axial muscles, or both,
-
may also have lesser involvement of oropharyngeal muscles.
Class IVb MG is characterized by the following:
-
predominantly affecting oropharyngeal, respiratory muscles or both,
-
may also have lesser or equal involvement of limb, axial muscles, or both.
Class V MG is characterized by the following:
-
intubation with or without mechanical ventilation, except when employed during routine postoperative management,
-
the use of feeding tube without intubation places the patient in class IVb.
Causes of Myasthenia Gravis
Nowadays the term ‘myasthenia gravis’ describes a heterogeneous group of autoimmune diseases with a postsynaptic defect of neuromuscular transmission. These myasthenic syndromes can be divided according to the following categories with distinct clinical features and specific therapeutic needs:
-
course type:
-
ocular (in approximately 20% of MG patients)
-
oropharyngeal or generalized
-
-
age of onset:
-
start before puberty
-
early onset before the age of 50 years
-
late onset after the age of 50 years [4]
-
-
antibody specificity:
-
anti-AChR
-
anti-muscle-specific receptor tyrosine kinase (MuSK)
-
anti-low-density lipoprotein receptor-related protein 4 (LRP4)
-
seronegative MG
-
-
pathology of the thymus
-
normal/atrophic thymus pathology
-
thymitis
-
paraneoplastic occurrence associated with thymoma
-
The immune system normally defends the body against diseases, but sometimes it can turn against the body, leading to an autoimmune disease. MG is just one of many autoimmune diseases, which include arthritis and type 1 diabetes.
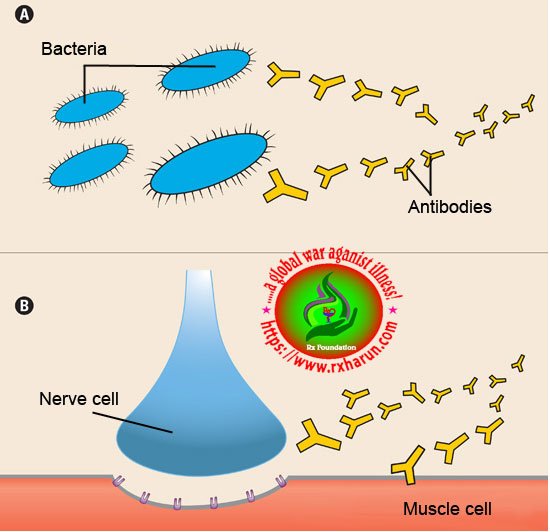
In all of these diseases, an army of immune cells that would normally attack bacteria and disease-causing “germs” mistakenly attacks cells and/or proteins that have essential functions in the body. In most cases of MG, the immune system targets the acetylcholine receptor — a protein on muscle cells that’s required for muscle contraction (see illustration to the right).
At the normal neuromuscular junction, a nerve cell tells a muscle cell to contract by releasing the chemical acetylcholine (ACh). ACh attaches to the ACh receptor — a pore or “channel” in the surface of the muscle cell — twisting it open and allowing an inward flux of electrical current that triggers muscle contraction. These contractions enable someone to move a hand, to dial the telephone, walk through a door or complete any other voluntary
movement.
About 85 percent of people with MG haveantibodies against the ACh receptor in their blood. The antibodies (Y-shaped missiles that immune cells called B cells use to attack bacteria and viruses) target and destroy many of the ACh receptors on muscle. Consequently, the muscle’s response to repeated nerve signals declines with time, and the muscles become weak and tired.
About 15 percent of people with MG areseronegative for antibodies to the ACh receptor, meaning the antibodies aren’t detectable in their blood (serum). Recently, it’s been discovered that a large fraction of these people have antibodies to muscle-specific kinase (MuSK), a protein that helps organize ACh receptors on the muscle cell surface.
Scientists don’t know what triggers most autoimmune reactions, but they have a few theories. One possibility is that certain viral or bacterial proteins mimic “self-proteins” in the body (such as the ACh receptor), stimulating the immune system to unwittingly attack the self-protein.
There’s also evidence that an immune system gland called the thymus plays a role in MG (see illustration below). Located in the chest just below the throat, the thymus is essential to the development of the immune system. From fetal life through childhood, the gland teaches immune cells called T cells to recognize self from non-self.

About 15 percent of people with MG have a thymic tumor, called a thymoma, and another 65 percent have overactive thymic cells, a condition calledthymic hyperplasia. When the thymus doesn’t work properly, the T cells might lose some of their ability to distinguish self from non-self, making them more likely to attack the body’s own cells.
What is the genetic susceptibility in MG?
Although MG and other autoimmune diseases are not hereditary, genetic susceptibility does appear to play a role.
Most studies suggest that if you have a relative with an autoimmune disease, your risk of getting an autoimmune disease is increased — the closer the relative, the higher the risk.
Even for identical twins, however, that risk is relatively small. Most studies suggest that when one twin has an autoimmune disease, the other has less than a 50 percent chance of getting the same disease.
Also, people who already have one autoimmune disease have a greater risk of developing another one. It’s estimated that 5 to 10 percent of people with MG have another autoimmune disease, which appeared before or after the onset of MG. The most common of these are autoimmune thyroid disease, rheumatoid arthritis and systemic lupus erythematosus (a disease that affects multiple organs).
Symptoms of Myasthenia Gravis

Muscle weakness caused by myasthenia gravis worsens as the affected muscle is used repeatedly. Because symptoms usually improve with rest, your muscle weakness may come and go. However, myasthenia gravis symptoms tend to progress over time, usually reaching their worst within a few years after the onset of the disease.
Although myasthenia gravis can affect any of the muscles that you control voluntarily, certain muscle groups are more commonly affected than others.
Eye muscles
In more than half the people who develop myasthenia gravis, their first signs and symptoms involve eye problems, such as:
- Drooping of one or both eyelids (ptosis).
- Double vision (diplopia), which may be horizontal or vertical, and improves or resolves when one eye is closed.
Face and throat muscles
In about 15 percent of people with myasthenia gravis, the first symptoms involve face and throat muscles, which can cause:
- Altered speaking – Your speech may sound very soft or nasal, depending upon which muscles have been affected.
- Difficulty swallowing – You may choke very easily, which makes it difficult to eat, drink or take pills. In some cases, liquids you’re trying to swallow may come out your nose.
- Problems chewing – The muscles used for chewing may wear out halfway through a meal, particularly if you’ve been eating something hard to chew, such as steak.
- Limited facial expressions – Your family members may comment that you’ve “lost your smile” if the muscles that control your facial expressions have been affected.
Neck and limb muscles
Myasthenia gravis can cause weakness in your neck, arms and legs, but this usually happens along with muscle weakness in other parts of your body, such as your eyes, face or throat.
The disorder usually affects arms more often than legs. However, if it affects your legs, you may waddle when you walk. If your neck is weak, it may be hard to hold up your head.
The presentation of MG has the following characteristics:
-
The usual initial complaint is a specific muscle weakness rather than generalized weakness
-
Extraocular muscle weakness or ptosis is present initially in 50% of patients and occurs during the course of illness in 90%
-
The disease remains exclusively ocular in only 16% of patients
-
Rarely, patients have generalized weakness without ocular muscle weakness
-
Bulbar muscle weakness is also common, along with weakness of head extension and flexion
-
Limb weakness may be more severe proximally than distally
-
Isolated limb muscle weakness is the presenting symptom in fewer than 10% of patients
-
Weakness is typically least severe in the morning and worsens as the day progresses
-
Weakness is increased by exertion and alleviated by rest
-
Weakness progresses from mild to more severe over weeks or months, with exacerbations and remissions
-
Weakness tends to spread from the ocular to facial to bulbar muscles and then to truncal and limb muscles
-
About 87% of patients have generalized disease within 13 months after onset
-
Less often, symptoms may remain limited to the extraocular and eyelid muscles for years
The following factors may trigger or worsen exacerbations:
-
Bright sunlight
-
Surgery
-
Immunization
-
Emotional stress
-
Menstruation
-
Intercurrent illness (eg, viral infection)
-
Medication (eg, aminoglycosides, ciprofloxacin, chloroquine, procaine, lithium, phenytoin, beta-blockers, procainamide, statins)
Diagnosis of Myasthenia Gravis
| Class | Description |
|---|---|
| I | Any eye muscle weakness, possible ptosis, no other evidence of muscle weakness elsewhere |
| II | Eye muscle weakness of any severity, mild weakness of other muscles |
| IIa | Predominantly limb or axial muscles |
| IIb | Predominantly bulbar and/or respiratory muscles |
| III | Eye muscle weakness of any severity, moderate weakness of other muscles |
| IIIa | Predominantly limb or axial muscles |
| IIIb | Predominantly bulbar and/or respiratory muscles |
| IV | Eye muscle weakness of any severity, severe weakness of other muscles |
| IVa | Predominantly limb or axial muscles |
| IVb | Predominantly bulbar and/or respiratory muscles |
| V | Intubation needed to maintain airway |
A doctor may perform or order several tests to confirm the diagnosis, including
- A physical and neurological examination – A physician will first review an individual’s medical history and conduct a physical examination. In a neurological examination, the physician will check muscle strength and tone, coordination, sense of touch, and look for impairment of eye movements.
- An edrophonium test – This test uses injections of edrophonium chloride to briefly relieve weakness in people with myasthenia gravis. The drug blocks the breakdown of acetylcholine and temporarily increases the levels of acetylcholine at the neuromuscular junction. It is usually used to test ocular muscle weakness.
- A blood test – Most individuals with myasthenia gravis have abnormally elevated levels of acetylcholine receptor antibodies. A second antibody—called the anti-MuSK antibody—has been found in about half of individuals with myasthenia gravis who do not have acetylcholine receptor antibodies. A blood test can also detect this antibody. However, in some individuals with myasthenia gravis, neither of these antibodies is present. These individuals are said to have seronegative (negative antibody) myasthenia.
- Electrodiagnostics – Diagnostic tests include repetitive nerve stimulation, which repeatedly stimulates a person’s nerves with small pulses of electricity to tire specific muscles. Muscle fibers in myasthenia gravis, as well as other neuromuscular disorders, do not respond as well to repeated electrical stimulation compared to muscles from normal individuals. Single fiber electromyography (EMG), considered the most sensitive test for myasthenia gravis, detects impaired nerve-to-muscle transmission. EMG can be very helpful in diagnosing mild cases of myasthenia gravis when other tests fail to demonstrate abnormalities.
- Diagnostic imaging – Diagnostic imaging of the chest using computed tomography (CT) or magnetic resonance imaging (MRI) may identify the presence of a thymoma.
- Pulmonary function testing – Measuring breathing strength can help predict if respiration may fail and lead to a myasthenic crisis.
Diagnostic tests
- Serum anti-acetylcholine receptor (ACh-R) antibody testing is the first-line investigation for non-urgent patients.
- Thyroid function for all patients.
- Serum MuSK antibody testing for all patients negative for ACh-R antibodies.
- Neurophysiological testing on symptomatic muscles may help to establish the diagnosis in seronegative patients with suspected MG. Repetitive nerve stimulation is the initial test. If this is negative then single-fibre electromyography should be considered.
- MR scan of brain: patients with negative serology and neurophysiology, and with symptoms compatible with ocular myasthenia, may have structural brain disease.
- Thymus CT or MRI scanning for all patients with suspected myasthenia, irrespective of distribution (ocular/generalised) or serology (seropositive/negative).
- Edrophonium test if there is diagnostic doubt – The edrophonium test involves intravenous administration of a short-acting acetylcholinesterase inhibitor while watching for a transient improvement in muscle strengt
Neurological examination
Your doctor may check your neurological health by testing your:
- Reflexes
- Muscle strength
- Muscle tone
- Senses of touch and sight
- Coordination
- Balance
- Breathing
- Seeing
- Swallowing
- Chewing
- Walking
- Using your arms or hands
- Holding up your head
The key sign that points to the possibility of myasthenia gravis is muscle weakness that improves with rest. Tests to help confirm the diagnosis may include:
Edrophonium test
- Injection of the chemical edrophonium chloride (Tensilon) may result in a sudden, although temporary, improvement in your muscle strength. This is an indication that you may have myasthenia gravis.
- Edrophonium chloride blocks an enzyme that breaks down acetylcholine, the chemical that transmits signals from your nerve endings to your muscle receptor sites.
Ice pack test
- If you have a droopy eyelid, your doctor may conduct an ice pack test. In this test, a doctor places a bag filled with ice on your eyelid. After two minutes, your doctor removes the bag and analyzes your droopy eyelid for signs of improvement. Doctors may conduct this test instead of the edrophonium test.
Blood analysis
- A blood test may reveal the presence of abnormal antibodies that disrupt the receptor sites where nerve impulses signal your muscles to move.
Repetitive nerve stimulation
- In this nerve conduction study, doctors attach electrodes to your skin over the muscles to be tested. Doctors send small pulses of electricity through the electrodes to measure the nerve’s ability to send a signal to your muscle.
- To diagnose myasthenia gravis, doctors will test the nerve many times to see if its ability to send signals worsens with fatigue.
Single-fiber electromyography (EMG)
- Electromyography (EMG) measures the electrical activity traveling between your brain and your muscle. It involves inserting a fine wire electrode through your skin and into a muscle to test a single muscle fiber.
Imaging scans
- Your doctor may order a CT scan or an MRI to check if there’s a tumor or other abnormality in your thymus.
Pulmonary function tests
- Your doctor may perform pulmonary function tests to evaluate whether your condition is affecting your breathing.
Treatment of Myasthenia Gravis
Today, myasthenia gravis can generally be controlled. There are several therapies available to help reduce and improve muscle weakness.
- Thymectomy – This operation to remove the thymus gland (which often is abnormal in individuals with myasthenia gravis) can reduce symptoms and may cure some people, possibly by rebalancing the immune system. A recent NINDS-funded study found that thymectomy is beneficial both for people with thymoma and those with no evidence of the tumors. The clinical trial followed 126 people with myasthenia gravis and no visible thymoma and found that the surgery reduced muscle weakness and the need for immunosuppressive drugs.
- Anticholinesterase medications – Medications to treat the disorder include anticholinesterase agents such as mestinon or pyridostigmine, which slow the breakdown of acetylcholine at the neuromuscular junction and thereby improve neuromuscular transmission and increase muscle strength.
Immunosuppressant drugs
- Cortroidsicoste – These drugs (which include prednisone and prednisolone) reproduce the actions of corticosteroid hormones, which are made by the cortex (outer layer) of the adrenal gland. They have broad anti-immune and anti-inflammatory effects, making them powerful treatments for MG. They’re not as fast-acting as cholinesterase inhibitors, but they’re faster than some other immunosuppressants, producing improvement within weeks to months. They’re also relatively inexpensive.
- A disadvantage of corticosteroids is that they can produce many side effects — some of them potentially serious — including osteoporosis (weakening of bones), mood disturbances, gastrointestinal problems, weight gain, high blood pressure, cataracts and stunted growth (in children). For many people, these side effects can be managed with other therapies; for example, bisphosphonate drugs can be used to prevent osteoporosis.
- Azathioprine (Imuran) – This was the first non-steroid immunosuppressant to come into widespread use against MG, in the 1970s. It acts more slowly than corticosteroids, producing improvement after three to six months, and usually has few side effects. Occasionally, however, it can produce serious side effects, including inflammation of the pancreas, liver toxicity, bone marrow suppression and possibly an increased risk of cancer.
- Mycophenylate mofetil (CellCept) – CellCept is a relatively new immunosuppressant, but so far it’s shown promising results against MG in clinical trials. In two small trials completed in 2001, about 65 percent of MG patients experienced gains in strength or a reduced need for prednisone after taking CellCept for several months. More recent analyses have shown that some people take longer to respond to the drug, but that nearly 75 percent eventually show improvement, with occasional relatively nonserious side effects such as stomach upset, flulike symptoms, rash and tremor.
- Cyclosporine (Neoral, Sandimmune) – This is a useful, relatively fast-acting treatment for MG, but it may have side effects including increased blood pressure, abnormal kidney function, unwanted body hair and stomach upset.
Cyclophosphamide (Cytoxan, Neosar) – This drug is considered effective against MG, but because it has many potentially serious side effects, it’s often reserved for use only when other drugs fail
- Plasmapheresis and intravenous immunoglobulin. These therapies may be options in severe cases of myasthenia gravis. Individuals can have antibodies in their plasma (a liquid component in blood) that attack the neuromuscular junction. These treatments remove the destructive antibodies, although their effectiveness usually only lasts for a few weeks to months.
- PyridostigmineThe first medicine tried is usually a tablet called pyridostigmine, which helps electrical signals travel between the nerves and muscles.It can reduce muscle weakness, but the effect only lasts a few hours so you’ll need to take it several times a day. For some people, this is the only medicine they need to control their symptoms.
- Intravenous immunoglobulin – is a highly concentrated injection of antibodies pooled from many healthy donors that temporarily changes the way the immune system operates. It works by binding to the antibodies that cause myasthenia gravis and removing them from circulation.
-
Plasma Exchange
Plasmapheresis is also known as a plasma exchange. This process removes harmful antibodies from the blood, which may result in an improvement in muscle strength.
Plasmapheresis is a short-term treatment. The body continues to produce the harmful antibodies and weakness may recur. Plasma exchange is helpful before surgery or during times of extreme MG weakness.
Myasthenia gravis facts
- Myasthenia gravis is a chronic autoimmune neuromuscular disease characterized by varying degrees of weakness of the skeletal (voluntary) muscles of the body.
- Myasthenia gravis is caused by a defect in the transmission of nerve impulses to muscles.
- The thymus may give incorrect instructions to developing immune cells, ultimately resulting in autoimmunity and the production of the acetylcholine receptor antibodies.
- The symptoms of myasthenia gravis may include eye muscle weakness, eyelid drooping (ptosis), blurry or double vision (diplopia), unstable gait, a change in facial expression, difficulty in swallowing, shortness of breath, impaired speech, and weakness in the arms, hands, fingers, legs, and neck.
- The disease is not directly inherited nor is it contagious; it commonly affects adult women (under 40) and older men (over 60), but it can occur at any age.
- Diagnosis is often delayed because muscle weakness is a common symptom in other diseases and may slowly develop; diagnostic tests that help confirm the diagnosis include detecting the presence of immune molecules or acetylcholine receptor antibodies, edrophonium test, and electromyography.
- Medical treatment includes anticholinesterase agents, plasmapheresis, and various immunosuppressive drugs; surgical treatment may include removal of the thymus.
- A myasthenic crisis occurs when the muscles that control breathing weaken so much that the patient requires emergency ventilation assistance.
- The disease prognosis is highly variable; some patients have complete remission (about 50% with thymus surgery), others have relatively normal lives with continued treatment, and others have a poor prognosis as the disease advances.
- Research is ongoing; new treatment protocols and immunosuppressive drugs are being investigated and therapeutic methods are likely to improve in the future.
|
Drugs that can exacerbate myasthenia gravis
|
|
|---|---|
| Drug group | Examples |
| Antibiotics | Aminoglycosides – eg, gentamicin Ciprofloxacin Macrolides – eg, erythromycin, azithromycin Tetracycline Ampicillin Clindamycin |
| Beta-blockers | Propranolol Atenolol Timolol eyedrops |
| Anti-arrhythmic drugs | Verapamil Quinidine and procainamide (both withdrawn) |
| Neuromuscular blocking agents | Atracurium Vecuronium(May cause unexpectedly long paralysis) |
| Other drugs | Lithium D-penicillamine Opiates – eg, pethidine Phenytoin Statins Magnesium Chloroquine Prednisolone |
Management of Myasthenia Gravis
Management of MG should be individualized according to patient characteristics and the severity of the disease. There are two approaches for the management of MG based on the pathophysiology of the disease. The first is by increasing the amount of Acetylcholine that is available to bind with the postsynaptic receptor using an acetylcholinesterase inhibitor agent, and the second is by using immunosuppressive medications that decrease the binding of acetylcholine receptors by antibodies.
There are four basic therapies used to treat MG
-
symptomatic treatment with acetylcholinesterase inhibitors,
-
rapid short-term immunomodulating treatment with plasmapheresis and intravenous immunoglobulin,
-
chronic long-term immunomodulating treatment with glucocorticoids and other immunosuppressive drugs,
-
surgical treatment.
Acetylcholinesterase Inhibitors
- Acetylcholinesterase inhibitors are the first-line treatment in patients with MG. Response to treatment varies from marked improvement in some patients to little or no improvement in others. Acetylcholinesterase inhibitors are used as a symptomatic therapy and act by increasing the amount of available acetylcholine at the NMJ [rx].
- They do not alter disease progression or outcome. Pyridostigmine is the most commonly used drug. It has a rapid onset of action within 15 to 30 minutes reaching peak activity in about two hours. The effect lasts for about three to four hours.
- The initial oral dose is 15–30 mg every 4–6 hours and is titrated upwards depending on the patient’s response. Adverse side effects of Pyridostigmine are mostly due to the cholinergic properties of the drug such as abdominal cramping, diarrhea, increased salivation and bronchial secretions, nausea, sweating, and bradycardia.
- Nicotinic side effects are also frequent and include muscle fasciculation and cramping. High doses of pyridostigmine exceeding 450 mg daily, administered to patients with renal failure, have been reported to cause worsening of muscle weakness [rx].
Short-Term Immunomodulating Therapies
- Plasma exchange and intravenous immunoglobulin have rapid onset of action with improvement within days, but this is a transient effect.
- They are used in certain situations such as myasthenic crisis and preoperatively before thymectomy or other surgical procedures. They can be used intermittently to maintain remission in patients with MG who are not well controlled despite the use of chronic immunomodulating drugs.
Plasmapheresis
- It improves strength in most patients with MG by directly removing AChR from the circulation [rx]. Typically one exchange is done every other day for a total of four to six times.
- Adverse effects of plasmapheresis include hypotension, paresthesias, infections, thrombotic complications related to venous access, and bleeding tendencies due to decreased coagulation factors [rx].
Intravenous Immunoglobulin Therapy (IVIg)
- It involves isolating immunoglobulins isolated from pooled human plasma by ethanol cryoprecipitation and is administered for 5 days at a dose of 0.4 g/kg/day, fewer infusions at higher doses are also used. The mechanism of action of IVIg is complex.
- Factors include inhibition of cytokines competition with autoantibodies and inhibition of complement deposition. Interference with the binding of Fc receptor on macrophages, Ig receptor on B cells, and interference with antigen recognition by sensitized T cells are other mechanisms [rx]. More specific techniques to remove pathogenic anti-AChR antibodies utilizing immunoadsorption have been developed recently, which offer a more targeted approach to MG treatment. Clinical trials showed a significant reduction of blocking antibodies with concomitant clinical improvement in patients treated with immunoadsorption techniques [rx].
- IVIg is considered to be safe but rare cases of complications do occur such as thrombosis due to increased blood viscosity and other complications related to large volumes of the infused preparation [rx].
- Compared to plasma exchange, IVIg is similar in terms of efficacy, mortality, and complications [rx]. However, plasma exchange (PLEX) has considerable cost advantages over IVIg with a cost-benefit ratio of 2 : 1 for treatment of myasthenia gravis [rx].
Long-Term Immune Therapies
- The goal of immune-directed therapy of MG is to induce remission or near remission of symptoms and maintain it.
Corticosteroids
- Corticosteroids were the first and most commonly used immunosuppressant medications in MG. Prednisone is generally used when symptoms of MG are not adequately controlled by cholinesterase inhibitors alone. A good response can be achieved with initial high doses and then tapering it to the lowest dose to maintain the response. A temporary exacerbation can occur after starting high doses of prednisone within the first 7–10 days which can last for several days [rx, rx].
- In mild cases, cholinesterase inhibitors are usually used to manage this worsening. In cases known to have severe exacerbations, plasma exchange or IVIg can be given before prednisone therapy to prevent or reduce the severity of corticosteroid-induced weakness and to induce a more rapid response. Oral prednisone might be more effective than anticholinesterase drugs in oMG and should therefore be considered in all patients with oMG [rx, rx].
Nonsteroidal Immunosuppressive Agents
- Azathioprine, a purine analog, reduces nucleic acid synthesis, thereby interfering with T-and B-cell proliferation. It has been utilized as an immunosuppressant agent in MG since the 1970s and is effective in 70%–90% of patients with MG [rx]. It usually takes up to 15 months to detect clinical response. When used in combination with prednisone, it might be more effective and better tolerated than prednisone alone [rx]. Adverse side effects include hepatotoxicity and leukopenia [rx].
- Mycophenolate mofetil selectively blocks purine synthesis, thereby suppressing both T-cell and B-cell proliferation. Widely used in the treatment of MG, its efficacy in MG was actually suggested by a few non-randomized clinical trials [rx, rx].
- The standard dose used in MG is 1000 mg twice daily, but doses up to 3000 mg daily can be used. Higher doses are associated with myelosuppression, and complete blood counts should be monitored at least once monthly. The drug is contraindicated in pregnancy and should be used with caution in renal diseases, GI diseases, bone marrow suppression, and elderly patients [rx].
- Cyclophosphamide administered intravenously and orally is an effective treatment for MG [rx]. More than half of the patients become asymptomatic within 1 year of treatment. Undesirable side effects include hair loss, nausea, vomiting, anorexia, and skin discoloration, which limit its use to the management of patients who do not respond to other immunosuppressive treatments [rx].
- Cyclosporine blocks the synthesis of IL-2 cytokine receptors and other proteins critical to the function of CD4+ T cells. Cyclosporin is used mainly in patients who do not tolerate or respond to azathioprine. Large retrospective studies have supported its use as a steroid-sparing agent [rx].
- Tacrolimus has been used successfully to treat MG at low doses. It has the theoretical advantage of less nephrotoxicity than cyclosporine. However, there are more controlled trial data supporting the use of cyclosporine. Like other immunosuppressive agents, Tacrolimus also has the potential for severe side effects [rx].
- MG patients resistant to therapy have been successfully treated with cyclophosphamide in combination with bone marrow transplant or with rituximab, a monoclonal antibody against the B cell surface marker CD20 [rx].
- Etanercept, a soluble and a recombinant tumor necrosis factor (TNF) receptor blocker, has also been shown to have steroid-sparing effects in studies on small groups of patients [rx, rx].
Drugs improving neuromuscular transmission: acetylcholinesterase inhibitors
- Pyridostigmine bromide is still the most commonly used acetylcholinesterase inhibitor in the treatment of MG, introduced more than 60 years ago [rx]. The administration of acetylcholinesterase inhibitors is symptomatic therapy without affecting the course of the disease with the risk of intoxication at very high daily doses (so-called ‘cholinergic crisis’).
- The daily dose of pyridostigmine bromide should exceed 600 mg only exceptionally. It is suitable as a long-term treatment in patients with very mild, non-progressive disease, and as adjunctive therapy in patients with severe disease who are also on immunotherapy.
- In general, however, pyridostigmine provides only limited benefit in severely affected patients. Pyridostigmine may cause bradycardia, especially in elderly patients. Increased bronchial and oral secretions are a serious problem in patients with swallowing or respiratory insufficiency. Patients on higher daily doses frequently complain about excessive sweating, muscle cramps and diarrhea.
- Switching medication to a sustained-release dosage form of pyridostigmine, which is available in only a limited number of countries (e.g. in the United States and Germany), may help to increase tolerability of this medication [rx].
- Anti-sense oligonucleotides present a novel type of AChE inhibition. AChE pre-mRNA is susceptible to alternative splicing. MG is associated with expression of the read-through transcript (AChE-R) which, unlike the normal ‘synaptic’ transcript (AChE-S), is not tethered to the post-synaptic membrane, but is a soluble monomer in the synaptic cleft.
- In experimental autoimmune MG (EAMG), inhibition of production of AChE-R using anti-sense oligonucleotides results in a significant reduction in synaptic expression of AChE-R. This improves muscle strength and increases the survival rates of experimental animals with EAMG [rx].
- In humans, there are only preliminary data on the therapeutic effect of Monarsen (EN101). This is a synthetic 20-base anti-sense oligodeoxynucleotide directed against the human AChE gene, modified to achieve stability for oral administration. Recent in-vitro and in-vivo studies indicate that EN101 is a Toll-like receptor (TLR)-9-specific ligand that can suppress proinflammatory functions and shift nuclear factor kappa B from the proinflammatory canonical pathway to the anti-inflammatory alternative pathway [rx]. TLR-9 is a member of the TLR family, which plays a fundamental role in pathogen recognition and activation of innate immunity.
Treatment of acute exacerbations
- Plasmapheresis, immunoadsorption and the intravenous administration of immunoglobulins, respectively, are used for crisis intervention. Only rarely do patients depend upon one of these therapies for a longer period of time [rx].
- Traditional plasma exchange entails removal of the pathogenic antibodies and other plasma components, such as soluble adhesion molecules and cytokines, separation from other blood components and then supplementation with 5% human albumin and crystalloids.
- The procedure may be carried out by plasma filtration techniques, plasma separation and more recently by antigen-specific immunoadsorption techniques that enable the return of non-pathogenic blood components to the patient. A standard course in MG entails five exchanges on alternating days utilizing 2–4 litres per exchange [rx].
- Venous access for plasma exchange can be achieved by central venous catheters or peripheral veins, and the preferred method varies among providers. Very recently, one retrospective study showed that peripheral veins access can be used successfully in most myasthenic patients and reduces the risk of serious and even lethal complications of the procedure [rx].
- A number of case reports and smaller, uncontrolled case series showed evidence for a roughly comparable clinical efficacy of plasmapheresis and immunoadsorption. However, the latter method avoids the necessity to substitute plasma replacement solution. This might result in better tolerability. Indeed, the first controlled study comparing the efficacy and safety of both treatments in myasthenic crisis confirms this advantage [rx].
- The use of high-dose intravenous immunoglobulin (IVIg) has gained wide application in the treatment of severe MG. Their mechanism of action is quite complex and not fully understood. IVIg seems to affect immune homeostasis by interfering at multiple levels, including modulation of the pathogenic autoantibody response, inhibition of complement activation and interference with the membrane attack complex formation, modulation of Fc receptors, down-regulation of the pathogenic cytokine responses and suppression of T cell function.
- The procedure usually entails the administration of 0·4 g/kg body weight human pooled IgG over 3 or 5 days [rx]. In acute exacerbations, including myasthenic crisis, intravenous Ig and plasma exchange have good and similar effects [rx,rx]. The major drawback of both is the relatively short-lived (in general up to 6 weeks) improvement in strength that makes the co-administration of longer acting immunosuppressive or immunomodulatory agents necessary.
Immunosuppressants
- Azathioprine still remains the first choice for long-term immunosuppressive therapy. However, it is important to point out that there are only very limited data from controlled studies on the efficacy of azathioprine [rx].
- A significant disadvantage of azathioprine is the delayed onset of action. Commonly, azathioprine is therefore started combined with prednisolone to achieve a rapid therapeutic effect.
- Individually adjusted to the patient’s needs, the prednisolone daily dose is then reduced gradually over a prolonged period of time. In a randomized double-blind study of 34 MG patients published in 1998, Palace et al. [rx] compared prednisolone and azathioprine versus prednisolone alone who were followed-up for 3 years. One group received prednisolone (on alternate days) plus azathioprine (2·5 mg/kg); the other group received prednisolone on alternate days plus placebo. Initial high-dose prednisolone (1·5 mg/kg on alternate days) was tapered at remission to the minimal dose required to maintain remission.
- The prednisolone dose did not differ significantly between the two groups at 1 year but was reduced at 2 and 3 years in the azathioprine group. Patients with refractory disease or azathioprine intolerance are dependent upon alternative corticosteroid sparing immunosuppressive treatment options than azathioprine. Many patients with generalized MG require lifelong immunosuppression.
- Corticosteroids are the least expensive, most reliable and rapid-acting drugs for immunomodulation in MG. This appraisal is based on general clinical experience rather than controlled studies. Their long-term use is complicated by severe and often intolerable adverse effects.
- The optimal manner to initiate corticosteroids depends upon the severity of weakness. It is important to note that with the initiation of higher doses, e.g. 60–100 mg prednisolone per day, some patients develop a significant exacerbation of myasthenic weakness, usually within the first day of treatment. The cause of this clinical phenomenon is elusive. In ocular MG a lower dose of prednisolone for a limited period of time, e.g. 20 mg for 2–4 weeks, can result in a pronounced improvement.
Cyclophosphamide
- Cyclophosphamide pulses (500–1000 mg/m2) given every 4–12 weeks has been used occasionally for refractory MG. Alternatively, it can also be given orally in a daily dose of 1–2 mg/kg body weight. Cyclophosphamide may be used only cautiously because of its myelotoxicity.
- Therefore, it is mandatory to evaluate again the necessity of the cyclophosphamide treatment after 6 months of treatment by omitting the medication or by tapering the daily dose, respectively. In pulse treatment, sufficient diuresis and the adjuvant mesna are required to reduce the risk of hemorrhagic cystitis.
- Additionally, myocardial damage, pulmonary fibrosis and cancer induction are possible consequences of the use of cyclophosphamide. Drachman et al. [rx] treated three myasthenic patients, for whom treatment with thymectomy, plasmapheresis and conventional immunosuppressive treatment failed, using high-dose cyclophosphamide (50 mg/kg/day intravenously for 4 days) followed by granulocyte colony-stimulating factor.
- It is known that such immunoablative treatment with high-dose cyclophosphamide does not damage hematopoietic stem cells, permitting repopulation of the immune system without bone marrow transplant. All three patients showed marked improvement in myasthenic weakness.
Methotrexate
- Methotrexate is a commonly used alternative to azathioprine. It is an anti-metabolite which has been used for decades in cancer therapy. In low doses, methotrexate is a generally safe and well-tolerated drug in the treatment of certain autoimmune diseases.
- However, there is only limited evidence for the effectiveness in MG. A recently published, single-blind study from South Africa provides evidence that methotrexate is an effective steroid-sparing agent 10 months after treatment initiation in generalized MG [rx]. This study suggests that methotrexate has similar efficacy and tolerability to azathioprine.
Mycophenolate mofetil
- Several retrospective case–series have suggested a very beneficial effect of this immunosuppressant. It is a reversible inhibitor of inosine monophosphate dehydrogenase in purine biosynthesis, which is necessary for T cells and B cells. Other cells are able to recover purines via a separate scavenger pathway and are thus able to escape the effect. Most patients well tolerate daily doses of 1–2·5 g. Two pivotal studies did not confirm the efficacy of mycophenolate in MG [rx,rx].
- However, it is important to note the relevant limitations of both studies. For example, the observation periods of 12 and 36 weeks, respectively, appear to be too short for an assessment of the drug effect in MG. In the open follow-up, patients on mycophenolate monotherapy began to improve between 6 and 12 months. In the combination therapy group, prednisone dose decreased after 12 months [rx].
- After more than 24 months, 53% were off prednisone and 75% took less than 7·5 mg prednisone per day. This follow-up corroborates the experience of previous retrospective and pilot studies in demonstrating that mycophenolate is an effective treatment for myasthenic patients as either monotherapy or adjunctive therapy to prednisone.
- The long follow-up demonstrated a steroid-sparing effect of mycophenolate during the second and third year of therapy that could not be demonstrated by studies of shorter duration. This illustrates that retrospective studies using rigorous outcomes measures can provide valuable information that may not be available from in short-term randomized controlled trials [rx].
Cyclosporin and tacrolimus
- Both are powerful immunosuppressive drugs used widely to prevent organ rejection after transplantation and in the treatment of autoimmune diseases. Although not structurally related to cyclosporin, tacrolimus has a similar mechanism of action and it has increasingly replaced cyclosporin for chronic immunosuppression after transplantation.
- Cyclosporin is a cyclic nonribosomal peptide of 11 amino acids and contains a single d-amino acid. Instead, tacrolimus has a macrolide lactone structure. Both inhibit the transcriptional activation of lymphokine and other genes required for T cell proliferation. The first step in mediating immunosuppressive effects is binding to their respective intracellular receptors, the immunophilin.
- The drug/immunophilin complex binds to and inhibits the calcium/calmodulin-dependent serine/threonine phosphatase calcineurin. Under normal circumstances, calcineurin is responsible for activating the transcription of interleukin 2. Possible risks of both drugs include nephrotoxicity, encephalopathy, hypertension and tremor.
- Cyclosporin is the immunosuppressive agent that revolutionized organ transplantation in the early 1980s by doubling the 1-year survival rate of cadaveric allografts. However, there are only scattered studies on the use of cyclosporin in MG [rx–rx]. In one controlled prospective and placebo-controlled study, 39 patients with steroid-dependent generalized MG received cyclosporin (5 mg/kg per body weight) or placebo for 6 months [rx].
- At the end of the study, patients in the cyclosporin group had significantly greater improvement in strength and a reduction in antibody titre. Percentage reduction of steroid medication was greater in the cyclosporin group, although the difference was not statistically significant.
- Cumulative side effects, however, caused a third of the patients to discontinue the medication; 10% did so secondary to slowly progressive nephrotoxicity. From our point of view, the limited evidence does not justify cyclosporin as a first-line immunosuppressant in MG. It is a reserve drug whose use is restricted due to serious side effects, numerous drug interactions, relatively high therapy costs and the necessity of regular measurements of the cyclosporin blood concentrations.
- Tacrolimus suppresses the induction of experimental autoimmune myasthenia in rats [rx]. In several small case–series tacrolimus lowered steroid requirements and induced stable remissions in MG [rx–rx].
- Recently, a double-blind, placebo-controlled, parallel group study focused on the ability of tacrolimus to reduce the corticosteroid dose in patients with stable myasthenic symptoms on prednisolone at doses equivalent to 10–20 mg/day [rx]. Patients received tacrolimus or placebo for 28 weeks and the corticosteroid dose was tapered with the procedures specified in the study protocol.
- Unfortunately, this short-duration trial provided a disappointing result regarding the primary efficacy end-point: the two study groups did not differ in the mean daily steroid dose. Besides immunosuppression, tacrolimus might have the potential to increase muscle strength by enhancing this ryanodine receptor function [rx]. However, it is doubtful if this effect is indeed of clinical significance.
Emerging therapy options
Rituximab
- The use of monoclonal antibodies with an innovative mode of action is promising and might change the treatment of MG significantly in the coming years. A particularly promising candidate is rituximab. This is a genetically engineered chimeric mouse/human monoclonal antibody representing a glycosylated immunoglobulin with human IgG1 constant regions and murine light-and heavy-chain variable region sequences [rx–rx].
- The antibody is produced by mammalian (Chinese hamster ovary) cell suspension culture. It is approved for the treatment of some lymphoma types and of severe active rheumatoid arthritis in adults who have had an inadequate response or intolerance to other disease modifying anti-rheumatic drugs.
- Rituximab binds specifically to the transmembrane antigen, CD20, a non-glycosylated phosphoprotein, located on pre-B and mature B lymphocytes. CD20 is found on both normal and malignant B cells, but not on hematopoietic stem cells, pro-B cells, normal plasma cells or other normal tissue.
- Toxicity studies have shown no other effect than the expected pharmacological depletion of B cells in peripheral blood and in lymphoid tissue. Peripheral B cell counts decline below normal following completion of the first dose of rituximab. B cell repletion begins within 6 months of treatment returning to normal levels between 9 and 12 months after completion of therapy [rx,rx].
- Rituximab provides promising expectations for the treatment of MG, although no randomized controlled trials have been conducted so far. Case reports, retrospective small series, and uncontrolled studies describe a long-lasting and pronounced clinical benefit due to rituximab even in severely affected, drug-resistant patients with MG [rx,rx–rx].
- Most interestingly, this might give better clinical benefit and last longer in anti-MuSK than in anti-AChR MG patients. Diaz-Manera et al. [22] treated six anti-MuSK-and 11 anti-AChR-positive MG patients with rituximab. All the patients included in this study were resistant to previous therapies and were classes III to V in the Myasthenia Gravis Foundation America classification.
- MuSK antibodies decreased dramatically during the follow-up after a single rituximab cycle. However, AChR antibodies remained at the same titers during the same period of time. After a mean post-treatment period of 31 months, 10 of the anti-AChR patients improved but six of them needed infusions. In contrast, all anti-MuSK patients achieved remission or minimal manifestations status and no infusions were needed. Consequently, in the anti-MuSK group, prednisone doses were reduced significantly and concomitant immunosuppressants could be withdrawn.
- Infusion reactions including fever, chills and shivering are the most common side effects of rituximab. Pretreatment with hydrocortisone and diphenhydramine ameliorates these reactions. Very rare cases of progressive multifocal leukoencephalopathy (PML) have been reported during the use of rituximab in lymphoma patients.
- The majority of patients suffering from this life-threatening complication had received rituximab in combination with chemotherapy. Cases of fatal PML have been reported following off-label use of rituximab for the treatment of certain autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis and vasculitis.
- These patients with autoimmune diseases had a history of prior or concurrent immunosuppressive therapy and were diagnosed with PML within 12 months of their last infusion of rituximab [rx–rx]. To date, there are no reports on the occurrence of PML in patients treated with rituximab for MG.
Bortezomib
- This is a proteasome inhibitor approved for treating patients with multiple myeloma, a plasma cell malignancy. Recent preclinical studies in cell cultures and animal models, and clinical studies in organ-transplant recipients, have demonstrated that bortezomib can kill non-neoplastic plasma cells within hours. This suggests that proteasome inhibitors could also be used for rapidly reducing autoantibody production in MG [rx].
Belimumab
- B cell activating factor (BAFF) is a potent B cell survival factor, and plays an essential role in B cell homeostasis and B cell function in the periphery [rx,rx]. Because BAFF is a crucial and potent factor for the survival and growth of B cells, both normal and autoreactive B cells compete for available BAFF.
- BAFF levels appear to regulate the survival threshold for B cells. Autoreactive B cells are poorly competitive for survival and they appear to be more dependent upon BAFF for their survival. BAFF levels are increased in the circulation of MG patients [rx].
- BAFF antagonists such as the monoclonal antibody belimumab may well provide new treatment options for MG. Belimumab is already approved in the United States, Canada and Europe for treatment of systemic lupus erythematosus.
Eculizumab
- Complement activation at the neuromuscular junction may be the primary cause of acetylcholine receptor loss and failure of neuromuscular transmission seen in MG. Eculizumab, a humanized monoclonal antibody, blocks the formation of terminal complement complex by selectively preventing the enzymatic cleavage of C5.
- It is the first therapy approved for the treatment of paroxysmal nocturnal hemoglobinuria. The results of one pilot Phase II study on the use of eculizumab in severe and refractory generalized MG are published in abstract form [rx].
- The purpose of this study was to determine whether or not eculizumab is safe and effective, despite treatment with various immunosuppressants that are currently available. The study has shown a significant clinical benefit of eculizumab in improving MG compared to placebo. Eculizumab was safe and well tolerated in all treated patients.
Etanercept
- Etanercept blocks tumour necrosis factor (TNF)-α activity. It has European approval to treat rheumatoid arthritis, juvenile rheumatoid arthritis and psoriatic arthritis, plaque psoriasis and ankylosing spondylitis. In animals, it suppresses ongoing experimental MG [rx].
- This observation resulted in a small prospective trial on the effect in corticosteroid-dependent MG. Eleven patients were enrolled, with eight completing this 6-month trial. Two patients were withdrawn owing to disease worsening, and one patient was withdrawn because of an erythematous skin rash.
- Six of the eight patients who completed the trial improved, based on quantitative measures of muscle strength and lowering of corticosteroid requirement [rx]. In addition to these disappointing study results, there are scattered case reports on patients who developed MG while taking etanercept and had a resolution of symptoms after stopping it [rx].
Granulocyte-macrophage colony-stimulating factor (GM-CSF)
- Forkhead box protein 3 (FoxP3) is a transcription factor necessary for the function of regulatory T cells (Tregs) [rx]. Tregs maintain immune homeostasis and self-tolerance and play an important role in the prevention of autoimmune disease. In-vitro administration of GM-CSF enhances the suppressive function of Tregs and up-regulated FoxP3 expression in Tregs.
- There is one single case report on a patient with a prolonged myasthenic crisis refractory to conventional immunomodulatory treatments who was treated with GM-CSF [rx]. This 77-year-old man received 750 μg GM-CSF daily for 3 days followed by 250 μg/day for 3 days. After the fifth dose of GM-CSF, he had improved generalized strength and was eventually weaned from the ventilator.
- The pillars of current MG therapy were already introduced more than 40 years ago. These are still pyridostigmine, corticosteroids, azathioprine and thymectomy. However, first experiences with new drugs, e.g. rituximab, are highly promising. It could be that the treatment of MG will change substantially until the end of this decade, but we are still far from a targeted immunotherapy.
Lifestyle & Home Remedies of Myasthenia Gravis
Supplementing your medical care with these approaches may help you make the most of your energy and cope with the symptoms of myasthenia gravis:
- Adjust your eating routine – Try to eat when you have good muscle strength. Take your time chewing your food, and take a break between bites of food. Small meals eaten several times a day may be easier to handle. Also, try eating mainly soft foods and avoid foods that require more chewing, such as raw fruits or vegetables.
- Use safety precautions at home – Install grab bars or railings in places where you may need support, such as next to the bathtub or next to steps. Keep your floors clean, and move any loose rugs out of areas where you may walk. Outside your home, keep paths, sidewalks, and driveways cleared of leaves, snow and other potential debris that could cause you to stumble.
- Use electric appliances and power tools – You may lose energy quickly when conducting tasks. Try using an electric toothbrush, electric can openers and other electrical tools to perform tasks when possible.
- Wear an eye patch – Consider wearing an eye patch if you have double vision, as this can help relieve the problem. Try wearing the eye patch while you write, read or watch television. Periodically switch the eye patch to the other eye to help reduce eyestrain.
- Plan – If you have chores, shopping or errands to do, plan the activity to coincide with the time at which you have the most energy. Also, try to reduce extra walking in your house when working on projects, as it may reduce your energy.
Supplemental & Physiotherapy for Myasthenia Gravis
MG can be fought with traditional medicine. But how else can you take a measure of control over your illness?
- Eat healthily – Like everyone, myasthenia patients should eat a healthy diet and maintain a healthy weight. This advice is critical for MG patients, because extra pounds make it even more fatiguing to get around and aggravate a host of other diseases. Talk to your doctor to see if a diet change will help ease medication side effects like fluid retention, bone loss or anemia.
- Exercise – It boosts your mood as well as the health of your heart, brain and cardiovascular system. Naturally, you have to tailor your activity to your illness. For example, when your disease is not well controlled, walking with assistance may be something to aim for. Exhaustion or shortness of breath means the exercise is too strenuous. As your MG improves, gradually add activity such as yoga, chair exercise, isometrics or more walking. Small progress is better than no progress!
- Manage stress -, Consult with your doctor and consider options such as acupuncture, biofeedback, meditation, or massage therapy.
- Get outdoors – Researchers have found that people can improve their mood with a short time of outdoor exercises like walking or gardening.
- Pretend you’re outdoors – Listening to recorded nature sounds can be very relaxing.
- Talk to others – Combat isolation by calling friends and family. Or contact a support group.
- Trust your spiritual side – For many, dealing with illness can be a conversation-starter with God.
References
