Chronic Myeloid Leukemia (CML) is a cancer of blood cells, characterized by replacement of the bone marrow with malignant, leukemic cells. Many of these leukemic cells can be found circulating in the blood and can cause enlargement of the spleen, liver, and other organs.
Chronic myelogenous leukemia, BCR–ABL1+ (CML) is a myeloproliferative neoplasm that originates in a pluripotent bone marrow stem cell and is consistently associated with the BCR–ABL1 fusion gene. This genetic abnormality results from the translocation of ABL1 on chromosome 9 to the region of the BCR gene on chromosome 22. The resulting fusion gene encodes an abnormal protein with constitutively activated tyrosine kinase activity that is responsible for the activation of signal transduction pathways that lead to the abnormal bone marrow proliferation and to the clinical and morphologic manifestations of this unique leukemia. Although the most prominent initial finding is usually leukocytosis owing to segmented neutrophils and their precursors at all stages of maturation, the BCR–ABL1 is found in all myeloid lineages and in some lymphoid and endothelial cells. The natural history of untreated CML is biphasic or triphasic: an initial indolent chronic phase (CP) is followed by an accelerated phase (AP), a blast phase (BP), or both.
Chronic Myeloid Leukaemia (CML) is a clonal, myeloproliferative disease that develops when a single, pluripotential, hemopoietic stem cell acquires the Philadelphia chromosome. CML was the first hematological malignancy to be associated with a specific genetic lesion
Chronic myeloid leukemia (CML), BCR-ABL1-positive, is classified as a myeloproliferative neoplasm predominantly composed of proliferating granulocytes and determined to have the Philadelphia chromosome/translocation t(9;22)(q34;q11.2). CML affects both the peripheral blood and the bone marrow.
Pathophysiology
The fusion oncoprotein BCR-ABL1 defines CML. 90% to 95% of patients with CML have a shortened chromosome 22 because of a reciprocal translocation t(9;22) (q34;q11.2) called the Philadelphia chromosome. The ABL1 gene encodes a non-receptor tyrosine kinase on chromosome 9, and BCR is a breakpoint cluster region on chromosome 22. The translated oncoprotein, in most cases, is 210-kd and called p210 BCR-ABL1. Alternative splicing results in p190 and p230 BCR-ABL1, which may show different presentations. This oncoprotein acts as a constitutively expressed defective tyrosine kinase. The downstream pathways affected include JAK/STAT, PI3K/AKT, and RAS/MEK; they involve cell growth, cell survival, inhibition of apoptosis, and activation of transcription factors.[rx]
The remainder of patients has variant or complex translocations involving additional chromosomes detected by routine cytogenetics or a cryptic BCR-ABL1 translocation detected with fluorescent in situ hybridization (FISH) or reverse transcriptase-polymerase chain reaction (PCR).[rx]
Types of Chronic Myeloid Leukemia
Classification
CML is often divided into three phases based on clinical characteristics and laboratory findings. In the absence of intervention, CML typically begins in the chronic phase, and over the course of several years progresses to an accelerated phase and ultimately to a blast crisis. Blast crisis is the terminal phase of CML and clinically behaves like acute leukemia. Drug treatment will usually stop this progression if started early. One of the drivers of the progression from chronic phase through acceleration and blast crisis is the acquisition of new chromosomal abnormalities (in addition to the Philadelphia chromosome).[rx] Some patients may already be in the accelerated phase or blast crisis by the time they are diagnosed.[rx]
Chronic phase
Approximately 85% of patients with CML are in the chronic phase at the time of diagnosis. During this phase, patients are usually asymptomatic or have only mild symptoms of fatigue, left side pain, joint and/or hip pain, or abdominal fullness. The duration of the chronic phase is variable and depends on how early the disease was diagnosed as well as the therapies used. In the absence of treatment, the disease progresses to an accelerated phase.[10] Precise patient staging based on clinical markers and personal genomic profile will likely prove beneficial in the assessment of disease history with respect to progression risk.[13]
Accelerated phase
Criteria for diagnosing transition into the accelerated phase are somewhat variable; the most widely used criteria are those put forward by investigators at M.D. Anderson Cancer Center,[rx] by Sokal et al.,[rx] and the World Health Organization.[rx][rx] The WHO criteria[rx] are perhaps most widely used, and define the accelerated phase by the presence of ≥1 of the following hematological/cytogenetic criteria or provisional criteria concerning response to tyrosine kinase inhibitor (TKI) therapy
Hematological/cytogenetic criteria
- Persistent or increasing high white blood cell count (> 10 × 109/L), unresponsive to therapy
- Persistent or increasing splenomegaly, unresponsive to therapy
- Persistent thrombocytosis (> 1000 × 109/L), unresponsive to therapy
- Persistent thrombocytopenia (< 100 × 109/L), unrelated to therapy
- ≥ 20% basophils in the peripheral blood
- 10―19% blasts in the peripheral blood and/or bone marrow
- Additional clonal chromosomal abnormalities in Philadelphia (Ph) chromosome-positive (Ph+) cells at diagnosis, including so-called major route abnormalities (a second Ph chromosome, trisomy 8, isochromosome 17q, trisomy 19), complex karyotype, and abnormalities of 3q26.2
- Any new clonal chromosomal abnormality in Ph+ cells that occurs during therapy
Provisional response-to-TKI criteria
- Hematological resistance (or failure to achieve a complete hematological response d) to the first TKI
- Any hematological, cytogenetic, or molecular indications of resistance to two sequential TKIs
- The occurrence of two or more mutations in the BCR-ABL1 fusion gene during TKI therapy
The patient is considered to be in the accelerated phase if any of the above are present. The accelerated phase is significant because it signals that the disease is progressing and transformation to blast crisis is imminent. Drug treatment often becomes less effective in advanced stages.[rx]
Blast crisis
Blast crisis is the final phase in the evolution of CML, and behaves like an acute leukemia, with rapid progression and short survival.[rx] Blast crisis is diagnosed if any of the following are present in a patient with CML:[rx]
- >20% blasts in the blood or bone marrow
- The presence of an extramedullary proliferation of blasts
Causes of Chronic Myeloid Leukemia
There is an increased incidence of CML among atomic bomb survivors; however, the predisposing risk factors are unknown.[rx]
Chronic myeloid leukemia is caused by a rearrangement (translocation) of genetic material between chromosome 9 and chromosome 22. This translocation, written as t(9;22), fuses part of the ABL1 gene from chromosome 9 with part of the BCR gene from chromosome 22, creating an abnormal fusion gene called BCR-ABL1. The abnormal chromosome 22, containing a piece of chromosome 9 and the fusion gene is often referred to as the Philadelphia chromosome (named for where it was first discovered). The translocation is acquired during a person’s lifetime and is present only in abnormal blood cells. This type of genetic change, called a somatic mutation, is not inherited.
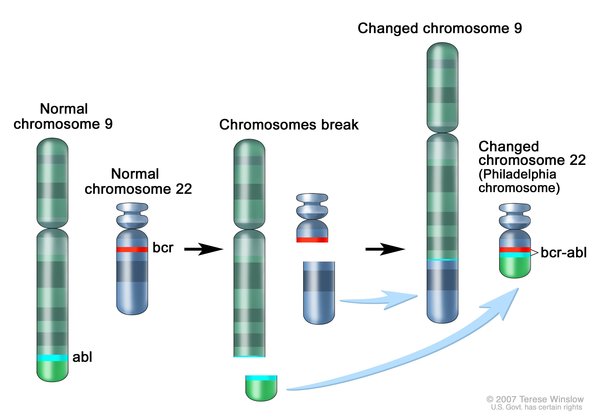{kind=link}
The function of the protein produced from the normal BCR gene is not completely understood, although it has been shown to help control signaling in cells. The protein produced from the normal ABL1 gene is involved in many cellular processes, including cell growth and division (proliferation), maturation (differentiation), movement (migration), and self-destruction (apoptosis).
Like the ABL1 protein, the abnormal protein produced from the fusion gene, called BCR-ABL1, can promote cell proliferation and block apoptosis. However, unlike ABL1, it does not require signals in the cell to turn it on. The constantly active BCR-ABL1 protein signals cells to continue dividing abnormally and prevents them from self-destructing, which leads to overproduction of the abnormal cells and, eventually, a shortage of normal blood cells. The presence of the Philadelphia chromosome provides a target for molecular therapies in people with chronic myeloid leukemia.
In 5 to 10 percent of cases of chronic myeloid leukemia, the BCR-ABL1 fusion gene is created by complex rearrangements that involve other chromosomes in addition to chromosomes 9 and 22. These genetic changes are called variant Philadelphia translocations. These cases are similar to those caused by t(9;22).
Researchers believe that additional genetic changes play a role in the progression of the chronic phase of chronic myeloid leukemia to the accelerated phase and blast crisis. The most common genetic changes associated with progression to blast crisis include an extra copy of chromosome 8 (trisomy 8), an abnormality of chromosome 17 known as isochromosome 17, and an extra copy (duplication) of the Philadelphia chromosome. When these somatic mutations occur in cells with the Philadelphia chromosome, they likely further promote uncontrolled cell proliferation.
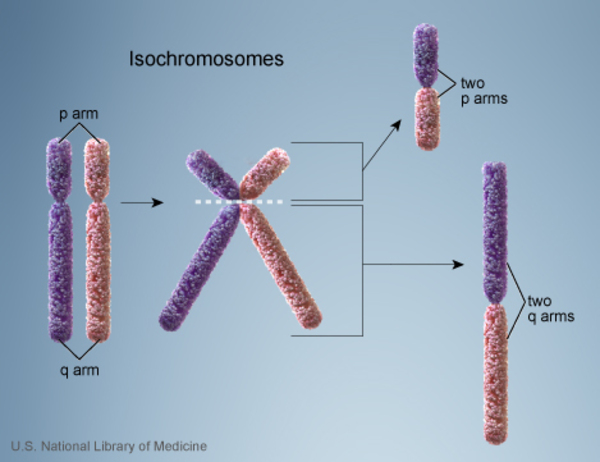{kind=link}
Chronic myelogenous leukemia occurs when something goes awry in the genes of your bone marrow cells. It’s not clear what initially sets off this process, but doctors have discovered how it progresses into chronic myelogenous leukemia.
An abnormal chromosome develops
Human cells normally contain 23 pairs of chromosomes. These chromosomes hold the DNA that contains the instructions (genes) that control the cells in your body. In people with chronic myelogenous leukemia, the chromosomes in the blood cells swap sections with each other. A section of chromosome 9 switches places with a section of chromosome 22, creating an extra-short chromosome 22 and an extra-long chromosome 9.
The extra-short chromosome 22 is called the Philadelphia chromosome, named for the city where it was discovered. The Philadelphia chromosome is present in the blood cells of 90 percent of people with chronic myelogenous leukemia.
The abnormal chromosome creates a new gene
The Philadelphia chromosome creates a new gene. Genes from chromosome 9 combine with genes from chromosome 22 to create a new gene called BCR-ABL. The BCR-ABL gene contains instructions that tell the abnormal blood cell to produce too much of a protein called tyrosine kinase. Tyrosine kinase promotes cancer by allowing certain blood cells to grow out of control.
The new gene allows too many diseased blood cells
Your blood cells originate in the bone marrow, a spongy material inside your bones. When your bone marrow functions normally, it produces immature cells (blood stem cells) in a controlled way. These cells then mature and specialize into the various types of blood cells that circulate in your body — red cells, white cells and platelets.
In chronic myelogenous leukemia, this process doesn’t work properly. The tyrosine kinase caused by the BCR-ABL gene allows too many white blood cells to grow. Most or all of these cells contain the abnormal Philadelphia chromosome. The diseased white blood cells don’t grow and die like normal cells. The diseased white blood cells build up in huge numbers, crowding out healthy blood cells and damaging the bone marrow.
Symptoms of Chronic Myeloid Leukemia
Chronic myelogenous leukemia often doesn’t cause signs and symptoms. It might be detected during a blood test.
When they occur, signs and symptoms may include:
- Bone pain
- Easy bleeding
- Feeling full after eating a small amount of food
- Feeling run-down or tired
- Fever
- Weight loss without trying
- Loss of appetite
- Pain or fullness below the ribs on the left side
- Excessive sweating during sleep (night sweats)
Accelerated phase
- Feel very tired
- Have a fever
- Get bruises
- Have night sweats
- Be short of breath
- Lose some weight
- Feel less hungry
- Get swelling or pain on your left side (which could be a sign of an enlarged spleen)
- Feel pain in your bones
Other symptoms may include stroke, changes in your vision, ringing in your ears, you feel like you’re in a daze, and you get prolonged erections.
Blastic phase
The leukemia cells multiply and crowd out healthy blood cells and platelets.
In this stage, you’ll have more severe symptoms, including:
- Infections
- Bleeding
- Skin changes including bumps, tumors
- Swollen glands
- Bone pain
Diagnosis of Chronic Myeloid Leukemia
Chronic Phase
The peripheral blood smear will show a leukocytosis due to granulocytes in various stages of maturation. There will be a bimodal distribution with higher proportions of mature segmented neutrophils and myelocytes. Blast cells will account for less than 2% of the white blood cells. Increased basophils and eosinophils are common. Significant dysplasia affecting greater than 10% of the granulocyte population is absent. Monocytosis may be present; however, it is most often less than 3% of the white blood cells. Platelets usually range from the normal range to a significant increase. Thrombocytopenia is an uncommon finding.[rx]
Bone marrow aspirate and biopsy will show hypercellularity with marked granulocytic proliferation and significantly increased myelocytes, although significant dysplasia should be absent. Blasts are usually less than 5%. Erythroid precursors are decreased considerably, and there is an increased myeloid to erythroid ratio. Megakaryocytes may be reduced, normal, or increased. About half of the cases show a megakaryocytic proliferation. The megakaryocytes in CML show a small, hypo-lobate “dwarf” morphology. The biopsy will show immature granulocytes in a thickened band of 5 to 10 cells along bone trabeculae. Adjacent to bone trabeculae is the normal distribution site of immature granulocytes; however, it is usually 2 to 3 cells thick. The bone marrow may also show increased reticulin fibrosis.[rx]
Accelerated Phase
The peripheral smear may or may not show increased blasts (10% to 19%). The bone marrow aspirate and biopsy will show similar changes to chronic phase CML with increased blasts (10% to 19%), possibly dysplastic changes in granulocytes, and increased reticulin and collagen fibrosis.[rx]
Blast Phase
The peripheral smear and/or bone marrow aspirate will show greater than 20% blasts, or there will be an extramedullary proliferation of blasts. Most cases will show blasts with myeloid differentiation; however, other lineages or combinations may be present, including lymphoblasts. Extramedullary proliferation is most commonly seen in skin, lymph nodes, bone, and the central nervous system (CNS).[rx]
History and Physical
Approximately half of the patients with CML are asymptomatic and are diagnosed on routine complete blood count. Most patients are in the chronic phase of CML. CML, in the chronic phase, most often presents with symptoms related to anemia and splenomegaly. Symptomatic anemia includes symptoms such as fatigue and malaise. Splenomegaly may cause a mass effect resulting in early satiety, left upper quadrant fullness, or pain. CML may also cause thrombocytopenia or platelet dysfunction resulting in bleeding, thrombocytosis resulting in thrombosis or priapism, basophilia resulting in histamine release and upper gastrointestinal ulcers. As CML progresses into the accelerated phase or blast phase, symptoms such as a headache, bone pain, fever, joint pain, bleeding, infections, and lymphadenopathy become more common.[rx]
The physical exam should include an assessment of the spleen size by palpation, measured in centimeters below the costal margin.[rx][rx] Splenomegaly is the most common physical exam finding in patients suffering from CML. In more than half of the patients, the spleen size extends beyond 5 cm below the left costal margin at the time of diagnosis. Avery large spleen is usually a herald sign of the transformation into an acute blast crisis from the disease. Hepatomegaly may also be found as it is a part of the extramedullary hematopoiesis occurring in the spleen. Physical exam findings may also include the signs of hyperviscosity. On fundoscopy, the retina may reveal papilledema, venous obstruction, and hemorrhages.
Evaluation
Initially, if CML is suspected, cytogenetic testing, fluorescent in situ hybridization (FISH), and/or reverse transcriptase-polymerase chain reaction (PCR) to determine the Philadelphia chromosome or BCR-ABL1 oncoprotein presence can be performed on peripheral blood.
At the time of diagnosis, laboratory blood testing should include a complete blood count with differential, chemistry panel, hepatitis panel, and a quantitative PCR for BCR-ABL1. A baseline bone marrow aspirate and biopsy should be performed with cytogenetics. Quantitative PCR should be repeated every three months after initiation of therapy. After BCR-ABL1 is less than or equal to 1% by international scale, quantitative PCR should continue for two years and then every 3 to 6 months after two years.
If chronic phase CML is established, additional evaluation includes determination of risk score using Sokal et al. or Hasford et al. risk calculations before determining first-line therapy.[rx]
Sokal risk calculation uses age, spleen size, platelet count, and percentage of myeloblasts in peripheral blood to determine the risk group.[rx]
Hasford risk calculation uses age, spleen size, platelet count, and percentage of blasts, eosinophils, and basophils in the peripheral blood to determine the risk group.[rx]
If accelerated or blast phase CML is diagnosed or progresses from chronic phase CML, additional testing should include flow cytometry to determine lineage, mutational analysis, and HLA testing if allogeneic hematopoietic stem cell transplant (HCT) is being considered.
Additional bone marrow cytogenetics and mutational analysis should be considered when there is a failure to reach response milestones or if there is any sign of hematologic or cytogenetic relapse.[rx]
- Complete blood count – It’s a blood test that checks to see how many white blood cells, red blood cells, and platelets you have.
- Bone marrow test – It helps you figure out how advanced your cancer is. Your doctor uses a needle to take a sample, usually from your hip bone.
FISH test (fluorescence in situ hybridization) – It’s a detailed lab test of your genes. - Ultrasound or CT scans – They can check the size of your spleen. Ultrasounds use sound waves to make images that doctors and other medical professionals can read. A CT is an X-ray that takes a series of pictures inside your body.
- Polymerase chain reaction test – It’s a lab test that looks for the BCR-ABL gene, which is involved in the process that tells your body to make too many of the wrong kind of white blood cells.
Treatment of Chronic Myeloid Leukemia
There are 4 FDA-approved, first-line treatments for chronic phase CML that are commercially available tyrosine kinase inhibitors, which include the first-generation imatinib and second-generation dasatinib, nilotinib, and bosutinib.
Dosing
FDA-approved and investigational BCR-ABL1 inhibitors targeting the kinase domain or the myristate pocket.
| Inhibitor | Binding site/ Inhibitor type |
Regulatory status/ approval |
|
|---|---|---|---|
| imatinib (Gleevec) | ATP-binding site/ATP-competitive | FDA approved/Frontline therapy | |
| nilotinib (Tasigna) | ATP-binding site/ATP-competitive | FDA approved/Frontline therapy | |
| dasatinib (Sprycel) | ATP-binding site/ATP-competitive | FDA approved/Frontline therapy | |
| bosutinib (Bosulif) | ATP-binding site/ATP-competitive | FDA approved/2nd-line therapy | |
| ponatinib (Iclusig) | ATP-binding site/ATP-competitive | FDA approved/2nd-line therapy | |
| ABL001 | Myristate pocket/Allosteric | Phase I/2nd-line therapy |
For chronic phase, CML with intermediate- or high-risk score, second-generation tyrosine kinase inhibitors (bosutinib, dasatinib, nilotinib) as first-line therapy may have an additional benefit over imatinib.[rx][rx][rx]
Ponatinib, a third-generation tyrosine kinase inhibitor, dosed at 45 mg daily, is a third-line treatment option in chronic phase CML for patients who have failed to respond to multiple tyrosine kinase inhibitors and for individuals who have the T315I mutation.[rx]
Advanced CML (accelerated or blast phase) has additional therapeutic considerations. Second- or third-generation tyrosine kinase inhibitor therapy should be initiated to reduce CML burden and be considered for early allogeneic hematopoietic stem cell transplant (HSCT).[rx] Omacetaxine is a chemotherapeutic agent that is an additional treatment option in cases refractory to tyrosine kinase inhibitor therapy that advanced from chronic phase CML.[rx]
Allogenic HSCT should be considered in patients resistant to tyrosine kinase inhibitor therapy in chronic phase CML and in the cases of advanced CML.[rx][rx]
Imatinib
A medicine called imatinib is now the main treatment for CML. It’s usually given soon after a diagnosis is made to slow the progression of the cancer and stop it reaching an advanced phase.
Imatinib works by reducing the production of abnormal white blood cells. It’s taken as a tablet once a day.
The side effects of imatinib are usually mild and should improve with time.
They can include:
- feeling and being sick
- swelling in the face and lower legs
- muscle cramps
- a rash
- diarrhea
Regular blood tests and occasional tests of your bone marrow will be carried out to check whether the treatment is working. If it does work, it will usually be taken for life.
Nilotinib
If you cannot take imatinib or it does not work for you, a medicine called nilotinib may be recommended instead. It’s also sometimes used as the first treatment for CML.
Nilotinib works in a similar way to imatinib and is taken as a capsule twice a day. If blood and bone marrow tests show the treatment is working, it’s also usually taken for life.
Common side effects of nilotinib include:
- headaches
- feeling sick
- abdominal (tummy) pain
- a rash
- itching
- hair loss
- muscle pain
- tiredness
If the side effects become particularly troublesome, temporarily stopping treatment usually helps to bring them under control. Treatment can then be resumed, possibly at a lower dose.
Dasatinib
If you cannot take imatinib or nilotinib, or they do not work for you, a similar medicine called dasatinib may be recommended.
This is taken as a tablet once a day and may be taken for life if blood and bone marrow tests show it’s working.
Side effects of dasatinib can include:
- an increased chance of picking up infections
- tiredness
- shortness of breath
- diarrhoea
- headaches
- a rash
Bosutinib
Bosutinib is a similar medication to imatinib, nilotinib and dasatinib. It may be recommended if you cannot take these medications, or you’ve tried them and they have not helped.
Bosutinib is taken as a tablet once a day and may be taken for life if blood and bone marrow tests show it is working.
Common side effects of bosutinib include:
- diarrhoea
- feeling and being sick
- abdominal pain
- a high temperature (fever)
- a rash
For more information, see the National Institute for Health and Care Excellence (NICE) guidelines on bosutinib for previously treated chronic myeloid leukaemia.
Ponatinib
Ponatinib is a similar medication to those mentioned above, but it’s only recommended for people with a specific genetic change (mutation) called the T315I mutation.
It’s taken as a tablet once a day and is taken for life if blood and bone marrow tests show it’s working.
Side effects of ponatinib can include:
- an increased risk of picking up infections
- tiredness
- shortness of breath
- headaches
- a rash
- aching joints
Combination therapy
In some cases, a combination of these medicines may be recommended. For example, a combination of high-dose imatinib, dasatinib and nilotinib may be recommended for people who did not respond to normal-dose imatinib.
Chemotherapy
Chemotherapy may be recommended if you cannot take the medicines above, or if CML has progressed to a more advanced stage. It may also be used while you’re awaiting tests results to confirm you have CML.
Chemotherapy involves taking medicine to kill cancerous cells. Tablets are usually used first because they have fewer and milder side effects than chemotherapy injections.
Side effects can include:
- tiredness
- a rash
- increased vulnerability to infection
If your symptoms persist or get worse, chemotherapy injections may be used. These have more side effects than tablets and they tend to be more severe.
In addition to the side effects mentioned above, side effects of chemotherapy injections can include:
- feeling and being sick
- hair loss
- infertility
The side effects should pass after your treatment has finished, although there’s a risk that infertility could be permanent.
Stem cell or bone marrow transplants
A stem cell or bone marrow transplant is the only potential cure for CML, but it’s a very intensive treatment and is not suitable for many people with the condition.
This is where donated cells called stem cells (which produce white blood cells) are transplanted into your body so you start to produce healthy white blood cells.
A stem cell transplant involves:
- having high-dose chemotherapy and radiotherapy to destroy the cancerous cells in your body
- removing stem cells from the blood or bone marrow of a donor – this will ideally be someone closely related to you, such as a sibling
- transplanting the donor stem cells into one of your veins
The high doses of chemotherapy and radiotherapy can put an enormous strain on the body and can cause significant side effects and life-threatening complications.
Transplants are generally only considered in younger people with CML, people in good general health and ideally those with a sibling who can provide a donation, as it’s more likely to be successful in these cases.
But in many cases of CML, the potential risks of transplantation far outweigh the potential benefits, particularly now that treatment with imatinib can often keep the condition under control for many years.
Bone marrow transplant
A bone marrow transplant also called a stem cell transplant, offers the only chance for a definitive cure for chronic myelogenous leukemia. However, it’s usually reserved for people who haven’t been helped by other treatments because bone marrow transplants have risks and carry a high rate of serious complications.
During a bone marrow transplant, high doses of chemotherapy drugs are used to kill the blood-forming cells in your bone marrow. Then blood stem cells from a donor are infused into your bloodstream. The new cells form new, healthy blood cells to replace the diseased cells.
Chemotherapy
Chemotherapy is a drug treatment that kills fast-growing cells in the body, including leukemia cells. Chemotherapy drugs are sometimes combined with targeted drug therapy to treat aggressive chronic myelogenous leukemia. Side effects of chemotherapy drugs depend on what drugs you take.
Immunotherapy
Immunotherapy is a treatment that uses the patient’s immune system to fight cancer. Substances made by the body or made in a laboratory are used to boost, direct, or restore the body’s natural defenses against cancer. This type of cancer treatment is also called biotherapy or biologic therapy.
High-dose chemotherapy with stem cell transplant
High doses of chemotherapy are given to kill cancer cells. Healthy cells, including blood-forming cells, are also destroyed by cancer treatment. A stem cell transplant is a treatment to replace the blood-forming cells. Stem cells (immature blood cells) are removed from the blood or bone marrow of the patient or a donor and are frozen and stored. After the patient completes chemotherapy, the stored stem cells are thawed and given back to the patient through an infusion. These reinfused stem cells grow into (and restore) the body’s blood cells.
Allogenic Stem Cell Transplants
Allogenic stem cell transplant (allo-SCT) has been used since the 1970s in the treatment of CML[rx] and is the only curative therapy for CML, however, it bears a significant mortality risk. Age, disease status, disease duration, recipient-donor gender combinations, degree of histocompatibility between donor and recipient, and the source of the transplant product have all been identified as significantly influencing long-term survival. Evidence in the pre-imatinib era suggests that bone marrow transplant is best performed in the early phase of chronic CML[rx],[rx]. Using blood or bone marrow-derived stem cells from an HLA-identical sibling performed in the chronic phase of the disease offers a 60-80% probability of leukemia-free survival at 5 years. If performed in the accelerated phase, disease survival decreases by half[rx].
Conventionally, conditioning treatments are necessary prior to all-SCT. This involves ‘myeloablative’ doses of chemoradiotherapy, aiming to facilitate engraftment of healthy donor stem cells via permanent elimination of malignant hematopoiesis. This is a rather arduous regimen associated with toxicity and mortality. It is therefore preferably administered to those aged less than 65 years without other co-morbid conditions. Success is generally attributed to an immunologically mediated graft-versus-leukemia effect[rx].
Bone marrow transplants have seen recent developments in research. Reduced-intensity conditioning treatments (RICT) or non-myeloablative transplants have been proposed. This endeavors to produce graft-versus-leukemia effects without exposing the patient to the potential toxicity of conditioning treatments. Here, the reconstitution of the immune system and associated anti-leukemia effect of the donor graft, compete against the growth of the malignancy. Preliminary data suggests that this approach may confer benefit, particularly in chronic phase CML[rx].
Interferon Alpha
Interferon-alpha (INFα), is a glycoprotein, of biological origin. It displays antiviral and antiproliferative properties. INFα was the first effective therapy for CML. The drug entered clinical trials in the early 1980s, and remained the treatment of choice for CML patients, until a shift in therapeutic strategy after the arrival of imatinib[rx]. In CML INFα prolongs survival in patients, especially of those who are cytogenetic responders. It is able to induce a cytogenetic response in 35 to 55% of patients, with a longer survival achievable in combination with chemotherapy. With this therapy, the level of disease decreased with time, but CML was rarely completely eliminated[rx].
Imatinib Mesylate
The BCR-ABL protein is an ideal drug target for CML treatment. Unique to leukemic cells, the BCR-ABL protein is expressed at high levels and its tyrosine kinase activity of the SH1 domain is essential for its ability to induce CML[rx]. The SH1 domain responsible for oncogenic transformation is an extremely attractive target in combating CML.
The most successful synthetic ATP inhibitor designed was imatinib mesylate (STI 571, Gleevec (Glivec), Novartis, Switzerland), approved by the Food and Drug Administration in May 2001 in the United States, later licensed for use in the UK by the European Medicines Evaluation Agency (EMEA) in November 2001 for the treatment of CML[rx,rx]. The introduction of this drug has dramatically changed the management of CML[rx. It is currently considered as the ‘gold standard’ in treating CML, approved for the first line treatment of adult patients with Ph+ CML at all disease stages[rx,rx].
Hommoharringtonine
Hommoharringtonine (HHT) is a novel plant alkaloid derived from a Chinese evergreen tree. An anticancer agent, it has recognized activity in acute myeloid leukemia (AML), having been incorporated into the treatment regimen for AML and CML[rx,rx]. HHT is thought to conduct its anti-leukemia effect through the inhibition of protein synthesis. HHT displays pronounced activity upon CML, in the past, it has been used as salvage therapy in patients who became refractory to INFα[rx]. Studies have investigated the consequences of HHT in combination with INFα or low dose cytarabine. When in dual therapy or in triple combination therapy, complete haematologic and complete cytogenetic responses equivalent to or superior to HHT single therapy have been shown, suggesting improved survival rates compared to HHT alone[rx–rx]. Shortly after such studies imatinib was introduced. In vitro HHT functions synergistically with imatinib, to decrease BCR-ABL protein expression. Research has shown imatinib and HHT to display synergistic cytotoxicity throughout different stages of disease progression. In the chronic phase, the duo demonstrated properties of dose-dependant apoptosis and growth inhibition[rx,rx]. Additional examination of the potential therapeutic effects of HHT as a single therapy or a dual regimen with imatinib is warranted.
Arsenic Trioxide
Arsenic trioxide (As2O3), an older therapy for CML, has been re-investigated. With the evolution of safer forms of arsenicals and efficacy of As2O3 in acute promyelocytic leukemia recently identified, the interest of its potential use in CML was rekindled[rx]. It is not certain how As2O3 exerts its anti-CML effects. Its ability to promote apoptosis has been suggested[rx]. Studies have shown dose-dependant growth inhibition and a pro-apoptotic effect when CML cells were treated with clinically tolerable levels of As2O3. A significant decline in BCR-ABL protein levels was also noted, and did not coincide with reduction in any other cellular proteins, suggesting specificity of this treatment. CML cell lines studies with As2O3 and imatinib have described a synergistic relationship between the two drugs, providing growth reduction and induction of apoptosis[rx,rx].
Other Novel therapies
Proteasome inhibition has been a further area of interest in CML therapy. The ubiquitin-proteasome pathway is responsible for the degradation of cellular proteins. Proteasome has a dual role of maintenance (disposal of damaged proteins) and regulation (degradation of proteins involved in cell cycle regulation and neoplastic growth) within the cell. Due particularly to its latter property, proteasome inhibitors are being investigated as new cancer therapy[rx]. The inactivation of NF-κB is pertinent to its action. Although the mechanism has not been established by which decreased expression of BCR-ABL protein is mediated when CML cells are treated with proteasome inhibitors; caspase activation and apoptosis were recognized. The proteasome inhibitor PS-341 has shown a significant effect upon growth inhibition and apoptosis of several cell lines. These have included both imatinib-resistant and sensitive BCR-ABL positive cell lines7. Again, clinical studies in imatinib-resistant patients are ongoing in this field[rx].
Pertinent Studies and Ongoing Trials
A search of ClinicalTrials.gov shows there are 221 active, interventional CML trials with 125 recruiting. These trials are widely variable with study topics covering novel drugs, treatment optimization of current treatment modalities, potential drug treatment combination regimens, the efficacy of treatment discontinuation, and immunotherapy.
Ongoing trials of note include the randomized international phase 3 study Evaluating Nilotinib Efficacy and Safety in Clinical Trials-Newly Diagnosed Patients (ENESTnd)[rx], and the phase 3 study Results of Efficacy and Safety of Radotinib Compared with Imatinib in Newly Diagnosed Chronic Phase Chronic Myeloid Leukemia (RERISE).[rx]
Other pertinent studies include trials that supported the first-line treatment with imatinib and dosing include the CML Study IV,[rx] the Tyrosine Kinase Inhibitor Optimization and Selectivity (TOPS) study,[rx] and the International Randomized Study of Interferon and STI571 trial (IRIS).[rx] Important trials concerning the efficacy and dosing of the second generation FDA-approved tyrosine kinase inhibitors include Bosutinib Efficacy and Safety in Newly Diagnosed CML trial (BELA),[rx] Bosutinib Versus Imatinib in Adult Patients With Newly Diagnosed Chronic Phase Chronic Myelogenous Leukemia trial (BFORE),[rx] Dasatinib Versus Imatinib Study in Treatment-Naive Chronic Myeloid Leukemia Patients trial (DASISION),[rx] and the ENESTnd study.[rx]
Pertinent studies and ongoing trials assessing tyrosine kinase inhibitor therapy discontinuation efficacy and candidate recommendations include the imatinib discontinuation study (STIM1),[rx] Evaluating Nilotinib Efficacy and Safety in Clinical Trials: Treatment-Free Remission study (ENESTfreedom),[rx] Imatinib Suspension and Validation study (ISAV),[rx] and Discontinuation of Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukaemia trial (EURO-SKI).[rx]
The European Society for Blood and Marrow Transplantation guidelines recommend allo-HSCT in the following cases:[rx]
- An EBMT risk score of 0–1 (the inclusion of second-generation TKI and transplantation are recommended after obtaining the optimal response);
- An EBMT risk score of 0–4 in cases of a loss of response to IM (hematological or cytogenetic). In patients with suboptimal response or failure of the first-line therapy in the following cases:
- In patients with no hematological response to second-generation TKI, regardless of the EBMT risk score (however, considering mutations within the BCR–ABL coding domain and previous allo-HSCT, these patients may benefit from treatment with third-generation TKI);
- In patients with failure to respond to IM therapies and second-generation TKI treatment-resistant BCR–ABL mutations as well as an EBMT risk score of 0–4;
- In patients in AP or BP after previous preparation with TKI or TKI in combination with polychemotherapy. Transplantation should be performed as soon as possible after reaching the second CP, without the need to achieve a deep cytogenetic or molecular response.
The use of the NCCN guidelines on the progression of the disease to the AP during first-line treatment with TKI has identical consequences. Second-line therapy with another TKI is considered a “bridging” treatment, which makes it possible to achieve an optimal response before HSCT. According to these guidelines, the application of omacetaxine is an alternative.[rx,rx]
Experts from Hammersmith Hospital in London presented a similar, somewhat more intuitive approach to the topic of qualifying patients to HSCT after failure of first-line therapy [rx]
Indications for allo-HSCT in chronic myeloid leukemia – the stance of experts from Hammersmith Hospital
| First chronic phase | Acceleration phase | Blast crisis phase | |
|---|---|---|---|
| Failure of the treatment with the available TKIs (donor search should be started after the failure of the first-line therapy) | Less advanced acceleration phase at the time of diagnosis – treatment as in the first chronic phase | Cases on the borderline of the blast phase and patients with the symptoms of transformation into the acceleration phase during treatment with TKI – treatment as in the case of the blast phase | HSCT immediately after obtaining the chronic phase using TKI or polychemotherapy (treatment with second-generation TKI after transplantation should be considered) |
Abbreviations: Allo-HSCT, allogeneic hematopoietic stem cell transplantation; TKI, tyrosine kinase inhibitor.
This brief analysis of treatment strategies in patients with advanced stages of the disease (AP and BP) indicates that this significantly differs from the treatment regimen of CP patients. More important is the unification of the AP and BP diagnostic criteria. The second important conclusion from this analysis relates to the effectiveness of AP and BP treatment with TKI. When IM is administered in the blast phase at a dose of 800 mg/day, complete hematological remission is achieved only in 25% of patients and 3-year PFS in 10%. In patients in the myeloid blast crisis (MBP) treated with dasatinib at 140 mg/day in one dose, a major cytogenetic response (MCyR) is achieved by 25% of individuals and by 28% of those receiving dasatinib in two doses (2×70 mg/day). In patients in the lymphoblastic crisis (LBP), MCyR is reached by 50% and 40%. Twenty-four-month overall survival (OS) is achieved by 24% and 28% of patients with MBP and 21% and 16% of those with LBP, respectively.[rx] These data have encouraged researchers to seek new molecular-oriented drugs that are an alternative to TKI.
Treatment of the second and subsequent lines is administered when the first-line therapy fails or the patient develops intolerance. The choice of the subsequent method of treatment is governed by additional rules:
- In the event of failure of first-line treatment with IM or its intolerance, we can administer dasatinib 100 mg/day, NIL 2×400 mg/day, or bosutinib 500 mg/day.
- In the event of failure of first-line treatment with dasatinib or its intolerance, we can administer NIL at a dose of 2×400 mg/day or bosutinib at a dose of 500 mg/day.
- In the event of failure of first-line treatment with NIL or its intolerance, we can administer dasatinib 100 mg/day or bosutinib 500 mg/day.
- Allo-HSCT is recommended only if first-line treatment with all available TKIs fails and the disease progresses to AP or BP, as well as in patients with the T315I mutation. This point does not administer to TKI intolerance. In subjects eligible for allo-HSCT, an attempt should be made to treat them with second-generation TKI and continue the treatment if: i) a minimal cytogenetic response (percentage of Ph+ cells <95%) is achieved after 3 months; ii) complete cytogenetic response (CCyR) is achieved after 6 months; and iii) major molecular response (MMR) is achieved after 1 year.
If this treatment is unsuccessful, allo-HSCT should be performed.
- In the case of failure of second-line treatment with dasatinib or its intolerance, third-line treatment can be administered with NIL at a dose of 2×400 mg/day, bosutinib at a dose of 500 mg/day, or ponatinib at a dose of 45 mg/day.
- In the case of failure of second-line treatment with NIL or its intolerance, in the third line we can administer dasatinib at a dose of 100 mg/day, bosutinib at a dose of 500 mg/day, or ponatinib at a dose of 45 mg/day.
- In the case of failure of second-line treatment with bosutinib or its intolerance, in the third-line therapy we can administer NIL 2×400 mg/day, dasatinib 100 mg/day, or ponatinib 45 mg/day.
- Allo-HSCT is recommended for all patients after failure of second-line therapy. This point does not apply to cases of intolerance to TKI.
- An attempt to treat with IM at a dose of 600–800 mg/day can be made in patients who are IM-resistant at a dose of 400 mg/day, have multiple comorbidities, are at high risk of second-generation TKI toxicity, and have good tolerance of IM.
Treatment Planning
Any of the 4 FDA-approved, tyrosine kinase inhibitors are highly effective in treating chronic phase CML. In the selection of front-line therapy, a patient’s age, comorbidities, and tyrosine kinase toxicities must be taken into account.
For patients with chronic phase CML, the use of the Sokal[rx] or Hasford[rx] risk stratification scores is helpful in predicting outcomes and choosing the first-line tyrosine-kinase inhibitor. For chronic phase, CML with intermediate- or high-risk score, second-generation tyrosine kinase inhibitors (bosutinib, dasatinib, nilotinib) may have the additional benefit over imatinib in achieving response milestones.[rx][rx][rx][rx]
For monitoring of treatment response, at baseline, all patients should undergo bone marrow examination and cytogenetic analysis. The patient should be monitored at 3, 6, and 12 months after starting therapy using cytogenetic and molecular studies such as PCR performed on peripheral blood.[rx] The major goal in chronic phase CML treatment is achieving complete cytogenetic response by 12 months after the initiation of tyrosine kinase inhibitors. Response milestones include complete hematologic response, cytogenetic response, and major molecular response.[rx] Complete hematologic response is defined as complete normalization of peripheral blood counts with a leukocyte count below 10 x 10^9/L and platelet count below 450 x 10^9/L, the absence of myelocytes, promyelocytes, and blasts, and absence of signs and symptoms of the disease including the disappearance of a palpable spleen.[rx] A complete cytogenetic response shows no Philadelphia-positive metaphases, and major molecular response is defined as BCR-ABL1 transcripts of less than or equal to 0.1% on the international scale (IS).[rx]
The European Leukemia Net (ELN) 2013 criteria can be a guide to monitor failure or suboptimal response to first-line therapy or second-line therapy. These criteria evaluate patients with chronic phase CML at 3 months, 6 months, and 12 months and then any subsequent follow-up; the optimal response is defined by PCR as BCR-ABL1 transcripts (IS) of less than or equal to 10%, less than 1%, and less than or equal to 0.1%, respectively. Alternatively, if quantitative PCR is not available, cytogenetics can be used to determine levels of the Philadelphia chromosome but is not useful in cryptic BCR-ABL1. If the optimal response is not achieved, there is a warning response group and a failure response group. In the warning group, consideration of dose escalation of imatinib or changing tyrosine kinase inhibitors is warranted.[rx] However, if an optimal response is not achieved at three months, studies show that most patients will achieve an optimal response at six months without changes in therapy with a similar prognosis.[rx] Another consideration is patient compliance; poor adherence has been shown to be a significant contributor to treatment failure and loss of complete response.[rx] In high-risk cases, at the time of diagnosis or in case of treatment failure despite good patient compliance, the tyrosine kinase inhibitor therapy should be changed, and the patient should be evaluated for the possibility of allogeneic HSCT.[rx]
In the event of tyrosine kinase inhibitor resistance, analysis of the BCR-ABL1 mutational profile may be useful in choosing second- or third-line therapy. The T315I mutation has been shown to confer resistance to imatinib and all second-generation tyrosine kinase inhibitors. In these patients, ponatinib should be considered.[rx] In cases of Y253H, E255K/V, or F359C/V mutations, dasatinib or bosutinib may have better responses, and in cases of V299L and F317L mutations, nilotinib may have promising outcomes.[rx]
For accelerated or blast phase CML, treatment considerations include evaluation for allogeneic HSCT, whether the advanced phase presented while the patient was on or off tyrosine kinase inhibitor therapy, comorbidities, age, prior therapy, and analysis of BCR-ABL1 mutation profile. Chemotherapy is often required in advanced CML to reduce disease burden and prepare for allogeneic HSCT.[rx][rx][rx]
In patients who achieve a long-term deep molecular response to therapy, discontinuation of tyrosine kinase inhibitors may be considered. Treatment-free remission can be achieved in approximately 40% to 60% of the low risk to intermediate-risk patients. Characteristics that predict treatment-free remission include cases with low Sokal risk, no history of tyrosine kinase inhibitor resistance, and duration of tyrosine kinase inhibitor use greater than five years. Most relapses will occur within the first six months of discontinuation, and molecular monitoring should occur monthly for the first year post-discontinuation and every 6 to 12 weeks indefinitely. If molecular relapse occurs, the tyrosine kinase inhibitor therapy should be re-initiated.[rx]
Blastic-Phase CML
Treatment Options for Blastic-Phase CML
-
Imatinib mesylate, dasatinib, and nilotinib have demonstrated activity in patients with myeloid blast crisis and lymphoid blast crisis or Philadelphia chromosome-positive acute lymphoblastic leukemia.[rx,rx] Two trials of imatinib mesylate and one trial of dasatinib involving a total of 518 patients in blastic-phase chronic myelogenous leukemia (CML) confirm a hematologic response rate of 42% to 55% and a major cytogenetic response rate of 16% to 25%, but the estimated 2-year survival rate is under 28%.[rx–rx][Level of evidence: 3iiiA] Clinical trials will explore combining imatinib mesylate with other drugs to improve the prognosis of patients with blastic-phase CML.[rx]
-
A review of 477 patients in the blastic phase CML treated between 1997 and 2016 at a single center showed that 72% had received previous tyrosine kinase inhibitor therapy in the chronic phase before the transformation.[rx] The median overall survival was 12 months and the median failure-free survival was 5 months.[rx][Level of evidence: 3iiiDiv] Patients who could complete an allogeneic stem cell transplant fared best, but this may have been due to selection bias.
-
Allogeneic bone marrow transplantation (BMT) represents the only potentially curative approach in these patients. Allogeneic BMT is more effective in patients induced into a second chronic phase.
-
Hydroxyurea as palliative therapy.
-
High-dose cytarabine.[rx]
Accelerated-Phase CML
Treatment Options for Accelerated-Phase CML
-
Allogeneic bone marrow transplantation or stem cell transplantation (SCT). In 132 patients with accelerated-phase chronic myelogenous leukemia (CML), a cohort study compared imatinib as first-line therapy with allogeneic SCT; with a median follow-up of 32 months, overall survival was improved using allogeneic SCT for the Sokal high-risk patients (100% vs. 17.7%; P = .008).[rx][Level of evidence: 3iiiA] Sokal low- and intermediate-risk patients showed no survival differences starting with either approach. Induction of remission using a tyrosine kinase inhibitor followed by an allogeneic SCT, when feasible, is a standard approach for patients with accelerated-phase CML.[rx]
-
Imatinib mesylate. Among 176 patients with accelerated-phase CML, the complete hematologic response was 82%, and the complete cytogenetic response was 43%; with a median follow-up of 41 months, the estimated 4-year survival was 53%.[rx] Other tyrosine kinase inhibitors need to be evaluated as first-line therapy in accelerated-phase CML.
-
Interferon-alpha.[rx] Although the response rate is lower for accelerated-phase disease than it is for chronic-phase disease, durable responses and suppression of cytogenetic clonal evolution have been reported.[rx,rx] When cytarabine was added to interferon-alpha, in comparison to historical controls of interferon alone, the response rate and 3-year survival appeared to be improved in late-stage patients.[rx][Level of evidence: 3iiiA]
-
High-dose cytarabine.[rx]
-
Hydroxyurea.
-
Busulfan.
-
Supportive transfusion therapy.
Patients with accelerated-phase CML show signs of progression without meeting the criteria for blast crisis (acute leukemia). Symptoms and findings include the following:
-
Increasing fatigue and malaise. (Refer to the PDQ summary on Fatigue for more information.)
-
Progressive splenomegaly.
-
Increasing leukocytosis and/or thrombocytosis.
-
Worsening anemia.
Bone marrow examination shows increasing blast cell percentage (but ≤30%) and basophilia. Additional cytogenetic abnormalities occur during the accelerated phase (trisomy 8, trisomy 19, isochromosome 17Q, p53 mutations or deletions), and the combination of hematologic progression plus additional cytogenetic abnormalities predicts for lower response rates and a shorter time-to-treatment failure on imatinib mesylate.[rx] At 1 year after the start of imatinib, the failure rate is 68% for patients with both hematologic progression and cytogenetic abnormalities, 31% for patients with only hematologic progression, and 0% for patients with cytogenetic abnormalities only. Before the availability of imatinib, the median survival time of accelerated-phase CML patients was less than 1 year.[rx]
Toxicity and Side Effect Management
Most patients will develop mild to moderate adverse events early in tyrosine kinase inhibitor therapy, and most will resolve spontaneously or will be well-controlled.
Adverse events and side effects can be divided into four grades based on severity.
-
Grade 1 – would require no change in tyrosine kinase inhibitor therapy; however, it may require specific treatment.
-
Grade 2 – will involve withholding therapy until severity decreases, or continuing therapy if symptoms decrease in severity with monitoring. If a Grade 2 side effect is recurrent, dose reduction should be considered.
-
Grade 3 – should involve withholding therapy until severity decreases and then restarting at a lower dose or withholding till symptoms reach a grade 1 level or less and resume prior dosage. If there is no resolution or Grade 3 side effects are recurrent, the tyrosine kinase inhibitor should be changed.
-
Grade 4 – events should involve switching tyrosine kinase inhibitor therapy when possible.[rx]
The side effects are
- Vascular System – Ischemic heart disease, ischemic cerebrovascular events, and peripheral arterial occlusive disease have been associated with nilotinib and ponatinib. Higher dosing has been related to higher risks of these events. Cardiovascular risk should be evaluated before therapy begins with these agents and should be closely monitored during therapy. Risk assessment and monitoring should include hemoglobin A1C, lipid profile, and serum creatinine. These drugs should be used with caution in patients with high risk for vascular disease.\
- Cardiac Function – Ponatinib has an associated incidence of heart failure. The monitoring of cardiac function should be mandatory for patients on ponatinib.
- Cardiac Rhythm Alterations – Tyrosine kinase inhibitors have the potential to alter the QT interval. Nilotinib has the highest incidence of QT interval prolongation. Ponatinib is associated with QT interval shortening. Electrocardiogram at baseline and periodically in select patients who are on any tyrosine kinase inhibitor and have QT interval prolongation, and for those who are on ponatinib. Nilotinib should be avoided in patients with high risk for arrhythmias or when other drugs that may alter the QT interval are being administered. Potassium and magnesium levels should be replete before initiation of tyrosine kinase inhibitor therapy in all cases.
- Pulmonary System – Pleural effusions can occur with all tyrosine kinase inhibitors, but is most commonly found with dasatinib. Patients with pre-existing lung injuries, chronic lung disease, congestive heart failure, and hypertension are at the highest risk for the development of pleural effusions. Caution should be taken if patients develop a cough, shortness of breath, or chest pain during therapy and a chest x-ray must be obtained in such cases. Pulmonary hypertension has also been reported with dasatinib use. If the development of pulmonary hypertension is suspected, dasatinib should be stopped immediately.
- Hepatobiliary System – Hepatotoxicity is common in all tyrosine kinase inhibitors. Ponatinib has the highest risk for developing high-grade increases in transaminases. Substances that alter liver metabolism by cytochrome P450 and acetaminophen should be used with caution or avoided entirely. Prompt treatment with glucocorticoids in severe cases of hepatotoxicity assists in hepatic recovery.
- Hyperglycemia – Nilotinib has been shown to induce hyperglycemia in non-diabetic and diabetic patients. Fasting glucose and hemoglobin A1C should be monitored.
- Hypercholesterolemia – Nilotinib has been associated with increases in LDL-cholesterol. Hypertriglyceridemia has been reported with ponatinib. If persistent high-risk hypercholesterolemia develops, appropriate statin initiation may be warranted.
- Other Endocrinopathies – Hypophosphatemia and hypocalcemia have been reported with imatinib, nilotinib, and dasatinib. Phosphate, calcium, and Vitamin D levels should be tested at baseline and monitored after that. Hypothyroidism may also develop and is treated effectively with hormone replacement.
- Myelosuppression – Development is common with all tyrosine kinase inhibitors, but is usually limited to the first few weeks to months of therapy. Leukocytes, erythrocytes, and platelets can all be affected. This necessitates close monitoring of blood counts and response to avoid infection and bleeding. Tyrosine kinase interruption, dose reduction, and supportive management with blood products are considerations based on levels of cytopenias, the specific tyrosine kinase inhibitor, and phase of the disease. Any nutritional deficiencies should be corrected before initiation of therapy.
- Gastrointestinal System – Nausea, diarrhea, abdominal pain, and vomiting are frequent side effects related to tyrosine kinase inhibitors. Diarrhea is especially significant with bosutinib and often requires dose interruption and/or dose reduction. To avoid nausea and vomiting, medication can be taken with meals. In severe cases, antiemetic and/or antidiarrheal medication can be administered, and close monitoring of hydration status should take place. Gastrointestinal bleeding has been reported with dasatinib. Increased lipase levels and pancreatitis have been reported with nilotinib and ponatinib.
- Dermatologic System – Rash development is common with all tyrosine kinase inhibitors. Most cases are dose-related and self-limited. Management can comprise topical therapies, antihistamines, and/or short courses of systemic steroids. In severe cases, prednisone can be considered with the gradual reintroduction of the offending tyrosine kinase inhibitor.
- Kidney Injury – Monitoring for tumor lysis syndrome should be done from the outset of treatment with all tyrosine kinase inhibitors. Otherwise, the development of acute kidney injury has been reported with imatinib therapy. Dasatinib and bosutinib have also been associated with acute kidney injury but at lower incidence rates. Serum creatinine should be monitored in these patients, and caution should be taken in patients with renal insufficiency.
Other common side effects that have been reported include myalgia, bone pain, ophthalmological changes, and headache.[rx][rx]
Staging
CML staging is determined by the phase of the disease, which includes a chronic phase, accelerated phase, and blast phase.
The chronic phase has less than 10% blasts, asymptomatic to mild symptoms, and responds to tyrosine kinase inhibitor therapy.
The World Health Organization (WHO) defines accelerated phase CML as having one or more of the following criteria:
-
Persistent or increasing high white blood cell count (greater than 10 x 10^9/L) unresponsive to therapy
-
Persistent or increasing splenomegaly unresponsive to therapy
-
Persistent thrombocytosis (greater than 1000 x 10^9/L) unresponsive to therapy
-
Persistent thrombocytopenia (less than 100 x 10^9/L)
-
Greater than or equal to 20% basophils in peripheral blood
-
10-19% blasts in peripheral blood or bone marrow (without extramedullary blast proliferation)
-
Additional chromosomal abnormalities in Philadelphia chromosome-positive cells at diagnosis
-
Any new clonal chromosomal abnormality in Philadelphia chromosome-positive cells during therapy
The WHO also considers resistance to tyrosine kinase inhibitors as provisional criteria for determining accelerated phase CML.
Blast phase CML is defined by greater than or equal to 20% blasts in the peripheral blood and/or bone marrow or extramedullary blast proliferation.[rx]
References